

Stratified analyses refine association between TLR7 rare variants and severe COVID-19
- PMID: 38944683
- PMCID: PMC11320601
- DOI: 10.1016/j.xhgg.2024.100323
Stratified analyses refine association between TLR7 rare variants and severe COVID-19
Abstract
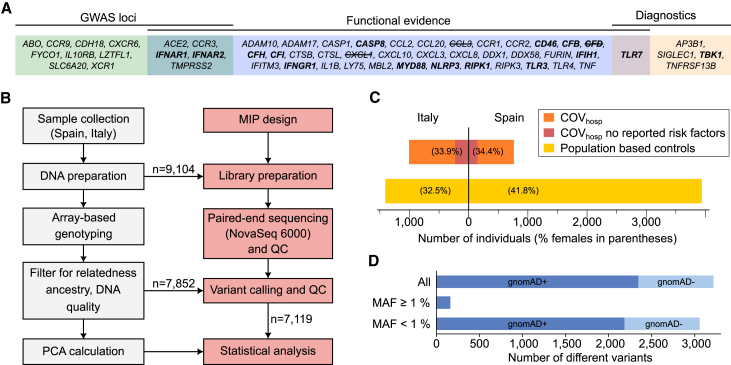
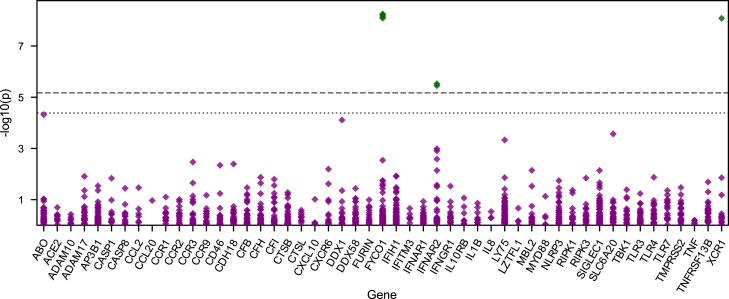
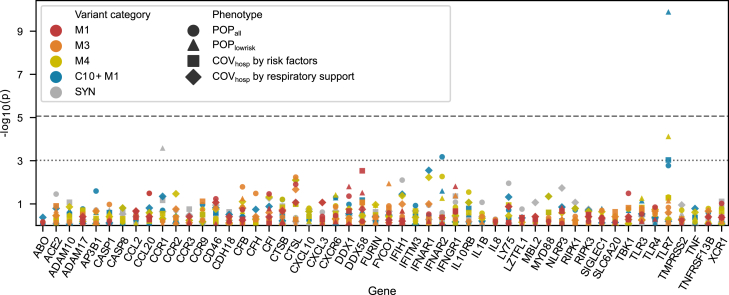
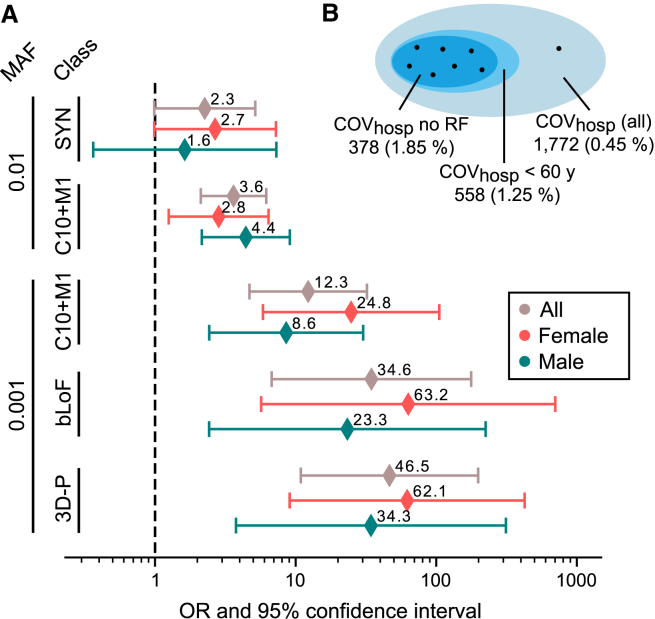
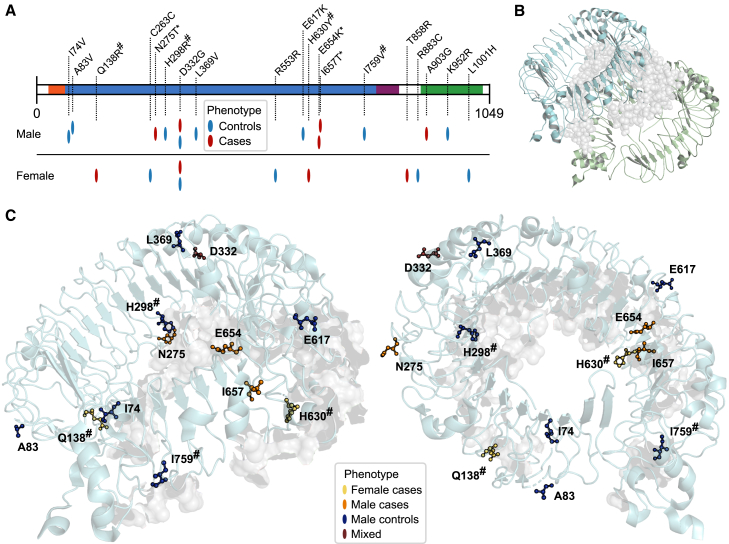
Despite extensive global research into genetic predisposition for severe COVID-19, knowledge on the role of rare host genetic variants and their relation to other risk factors remains limited. Here, 52 genes with prior etiological evidence were sequenced in 1,772 severe COVID-19 cases and 5,347 population-based controls from Spain/Italy. Rare deleterious TLR7 variants were present in 2.4% of young (<60 years) cases with no reported clinical risk factors (n = 378), compared to 0.24% of controls (odds ratio [OR] = 12.3, p = 1.27 × 10-10). Incorporation of the results of either functional assays or protein modeling led to a pronounced increase in effect size (ORmax = 46.5, p = 1.74 × 10-15). Association signals for the X-chromosomal gene TLR7 were also detected in the female-only subgroup, suggesting the existence of additional mechanisms beyond X-linked recessive inheritance in males. Additionally, supporting evidence was generated for a contribution to severe COVID-19 of the previously implicated genes IFNAR2, IFIH1, and TBK1. Our results refine the genetic contribution of rare TLR7 variants to severe COVID-19 and strengthen evidence for the etiological relevance of genes in the interferon signaling pathway.
Keywords: SARS-CoV-2; burden analysis; host genetics; immune deficiency; infection; innate immunity; rare variants; targeted sequencing; toll-like receptor 7; variant collapsing analysis.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.U.L. is a co-founder of LAMPseq Diagnostics GmbH.
Figures
References
-
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet Lond. Engl. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. - DOI - PMC - PubMed
-
- Bastard P., Gervais A., Le Voyer T., Rosain J., Philippot Q., Manry J., Michailidis E., Hoffmann H.-H., Eto S., Garcia-Prat M., et al. Autoantibodies neutralizing type I IFNs are present in ∼4% of uninfected individuals over 70 years old and account for ∼20% of COVID-19 deaths. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abl4340. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
