

This is a preprint.
Variants in the Kallikrein Gene Family and Hypermobile Ehlers-Danlos Syndrome
- PMID: 38947032
- PMCID: PMC11213194
- DOI: 10.21203/rs.3.rs-4547888/v1
Variants in the Kallikrein Gene Family and Hypermobile Ehlers-Danlos Syndrome
Abstract
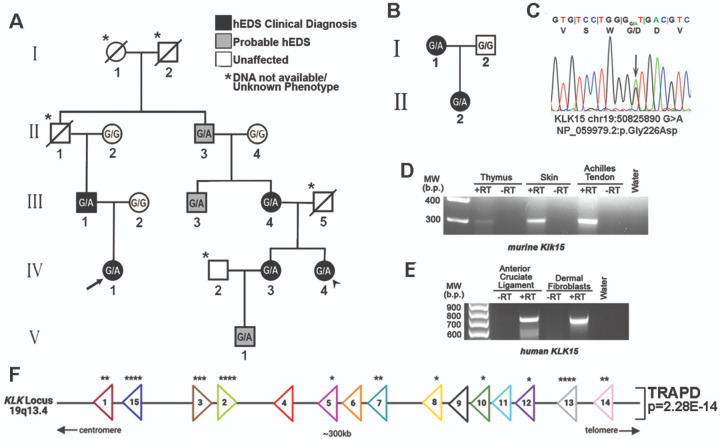
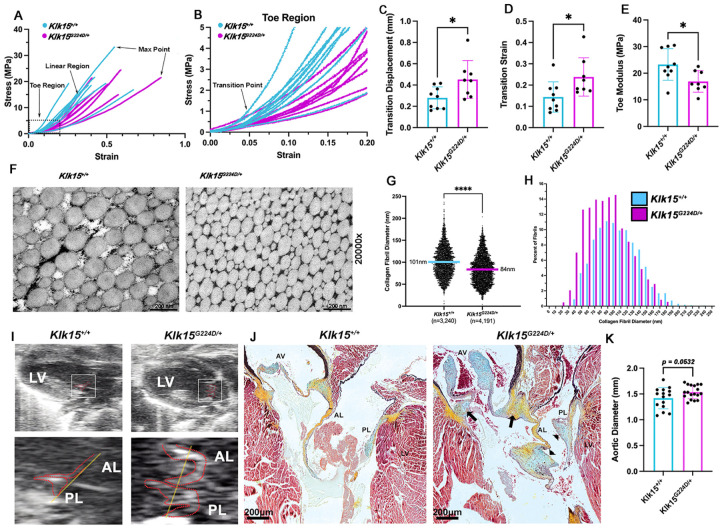
Hypermobile Ehlers-Danlos syndrome (hEDS) is a common heritable connective tissue disorder that lacks a known genetic etiology. To identify genetic contributions to hEDS, whole exome sequencing was performed on families and a cohort of sporadic hEDS patients. A missense variant in Kallikrein-15 (KLK15 p. Gly226Asp), segregated with disease in two families and genetic burden analyses of 197 sporadic hEDS patients revealed enrichment of variants within the Kallikrein gene family. To validate pathogenicity, the variant identified in familial studies was used to generate knock-in mice. Consistent with our clinical cohort, Klk15 G224D/+ mice displayed structural and functional connective tissue defects within multiple organ systems. These findings support Kallikrein gene variants in the pathogenesis of hEDS and represent an important step towards earlier diagnosis and better clinical outcomes.
Keywords: Connective tissues; Hypermobile Ehlers Danlos Syndrome; hEDS.
Figures
References
-
- Petrucci T, B.J., Gensemer C, Morningstar J, Daylor V, Byerly K, Bistran E, Griggs M, Elliott JM, Kalechi T, Phillips S, Nichols M, Shapiro S, Patel S, Bouatia-Naji N, Norris RA Phenotypic Clusters and Multimorbidity in Hypermobile Ehlers-Danlos Syndrome. Mayo Clinic Proceedings: Innovations, Quality & Outcomes 8, 253–262 (2024). - PMC - PubMed
Publication types
Grants and funding
- P20 GM121342/GM/NIGMS NIH HHS/United States
- K99 CA207729/CA/NCI NIH HHS/United States
- T32 GM132055/GM/NIGMS NIH HHS/United States
- P20 GM103444/GM/NIGMS NIH HHS/United States
- R01 HL131546/HL/NHLBI NIH HHS/United States
- UL1 TR004419/TR/NCATS NIH HHS/United States
- R01 DE021134/DE/NIDCR NIH HHS/United States
- T32 HL007260/HL/NHLBI NIH HHS/United States
- U01 DE031512/DE/NIDCR NIH HHS/United States
- R01 HL149696/HL/NHLBI NIH HHS/United States
- R00 CA207729/CA/NCI NIH HHS/United States
- F31 HL167482/HL/NHLBI NIH HHS/United States
- C06 RR018823/RR/NCRR NIH HHS/United States
- F31 HL158243/HL/NHLBI NIH HHS/United States
- F32 AI181339/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
