

This is a preprint.
Characterization of the family-level Borreliaceae pan-genome and development of an episomal typing protocol
- PMID: 38947078
- PMCID: PMC11213207
- DOI: 10.21203/rs.3.rs-4491589/v1
Characterization of the family-level Borreliaceae pan-genome and development of an episomal typing protocol
Update in
-
Characterization of the family-level Borreliaceae pan-genome and development of an episomal typing protocol.mBio. 2025 Jun 11;16(6):e0094325. doi: 10.1128/mbio.00943-25. Epub 2025 May 7. mBio. 2025. PMID: 40331826 Free PMC article.
Abstract
Background: The Borreliaceae family includes many obligate parasitic bacterial species which are etiologically associated with a myriad of zoonotic borrelioses including Lyme disease and vector-borne relapsing fevers. Infections by the Borreliaceae are difficult to detect by both direct and indirect methods, often leading to delayed and missed diagnoses. Efforts to improve diagnoses center around the development of molecular diagnostics (MDx), but due to deep tissue sequestration of the causative spirochaetes and the lack of persistent bacteremias, even MDx assays suffer from a lack of sensitivity. Additionally, the highly extensive genomic heterogeneity among isolates, even within the same species, contributes to the lack of assay sensitivity as single target assays cannot provide universal coverage. This within-species heterogeneity is partly due to differences in replicon repertoires and genomic structures that have likely arisen to support the complex Borreliaceae lifecycle in which these parasites have to survive in multiple hosts each with unique immune responses.
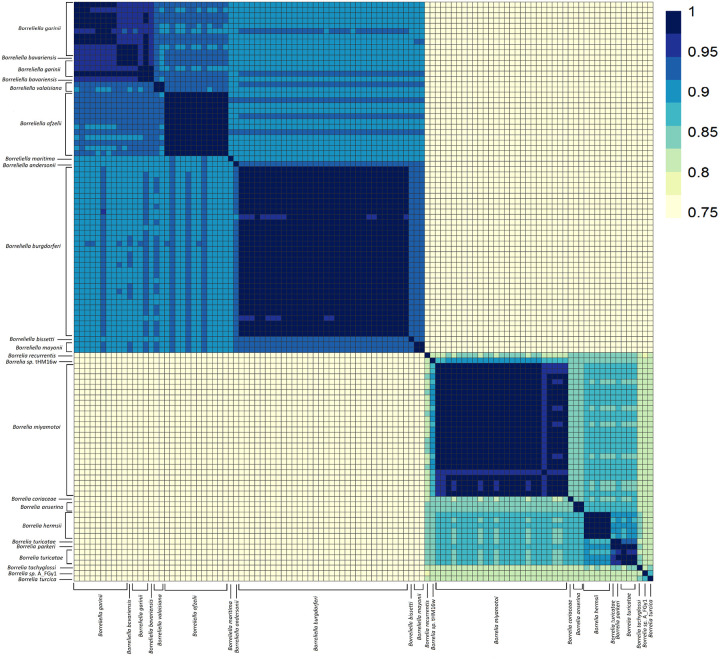
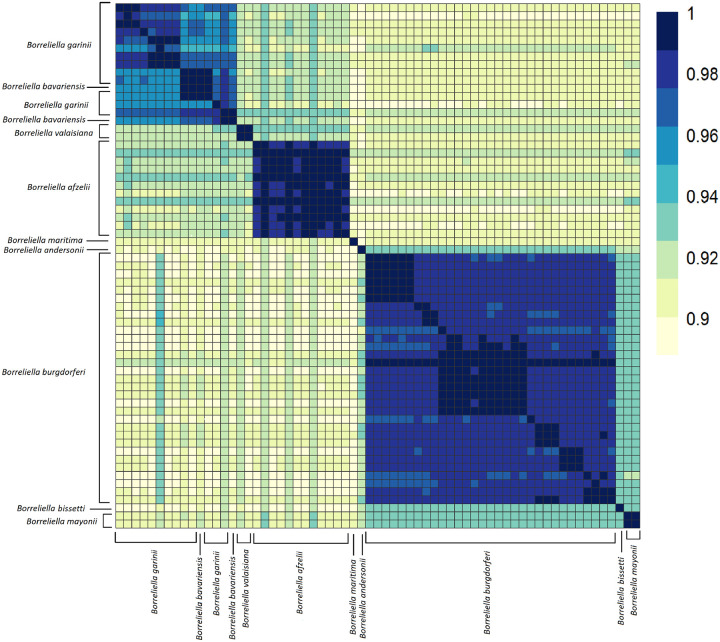
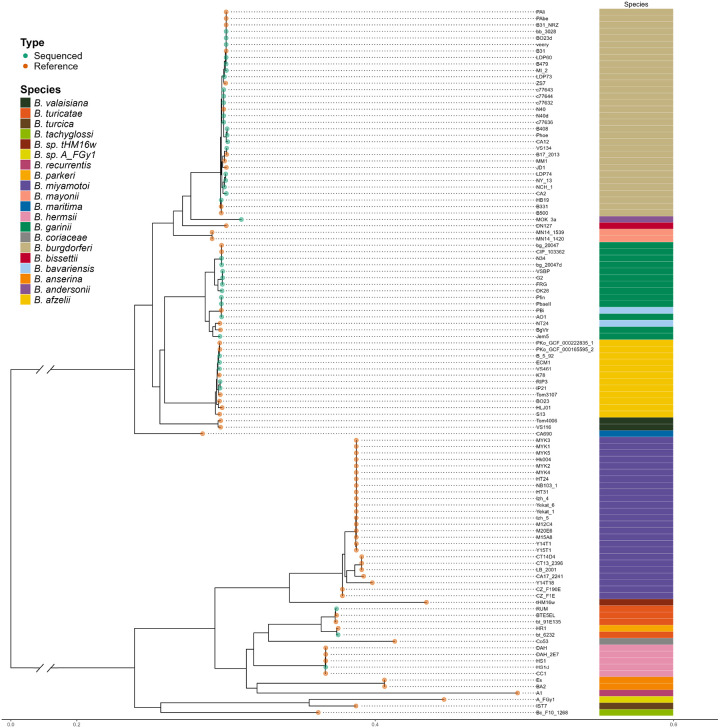
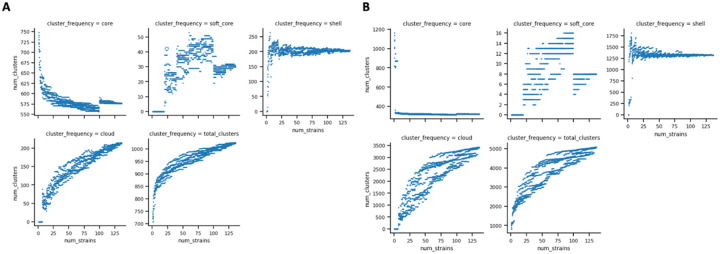
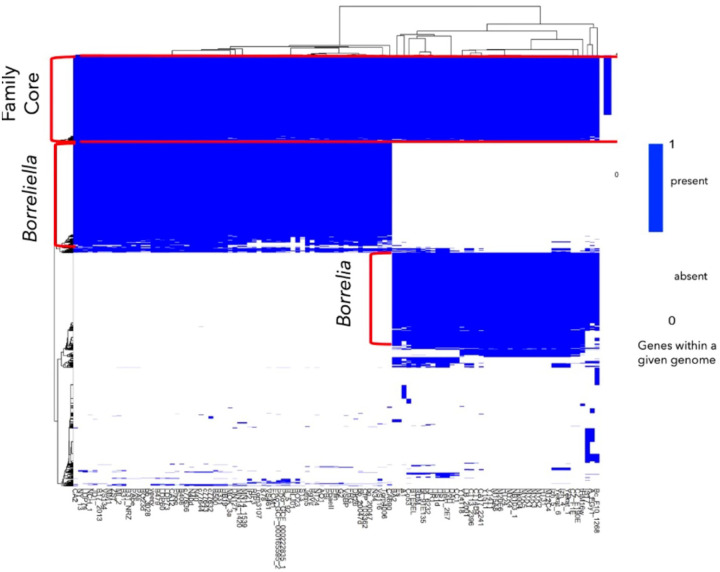
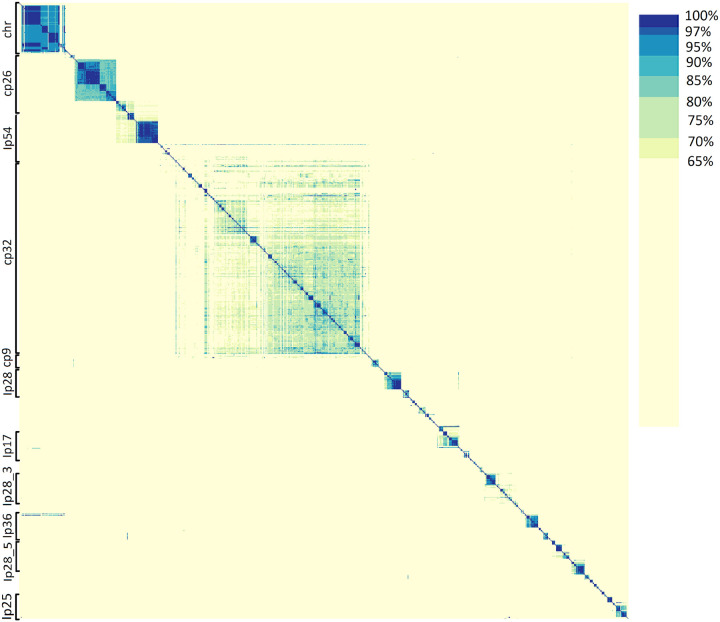
Results: We constructed a Borreliaceae family-level pangenome and characterized the phylogenetic relationships among the constituent taxa which supports the recent taxonomy of splitting the family into at least two genera. Gene content pro les were created for the majority of the Borreliaceae replicons, providing for the first time their unambiguous molecular typing.
Conclusion: Our characterization of the Borreliaceae pan-genome supports the splitting of the former Borrelia genus into two genera and provides for the phylogenetic placement of several non-species designated isolates. Mining this family-level pangenome will enable precision diagnostics corresponding to gene content-driven clinical outcomes while also providing targets for interventions.
Keywords: Borrelia; Borreliaceae; Borreliella; Spirochete; comparative genomics; distributed genome hypothesis; pan-genome; phylogenetics; supragenome; taxonomy.
Conflict of interest statement
Competing interests (NONE) Additional Declarations: No competing interests reported.
Figures




References
-
- Shen K, Wang X, Post JC, Ehrlich GD. Molecular and Translational Research Approaches for the study of Bacterial Pathogenesis in Otitis Media. In: Rosenfeld R, Bluestone CD, editors. Evidence-based Otitis Media. 2nd ed. Hamilton, London: B.C. Decker Inc.; 2003. pp. 91–119.
-
- Ehrlich GD, Hu ZF, Post JC. Role for Biofilms in Infectious Disease. In: Ghannoum M, O’Toole GA, editors. Microbial Biofilms. Washington, D.C.: ASM; 2004. pp. 332–58.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
