

This is a preprint.
A genome resource for the marine annelid Platynereis dumerilii
- PMID: 38948846
- PMCID: PMC11213123
- DOI: 10.1101/2024.06.21.600153
A genome resource for the marine annelid Platynereis dumerilii
Update in
-
A genome resource for the marine annelid Platynereis spp.BMC Genomics. 2025 Jul 14;26(1):665. doi: 10.1186/s12864-025-11727-2. BMC Genomics. 2025. PMID: 40660120 Free PMC article.
Abstract
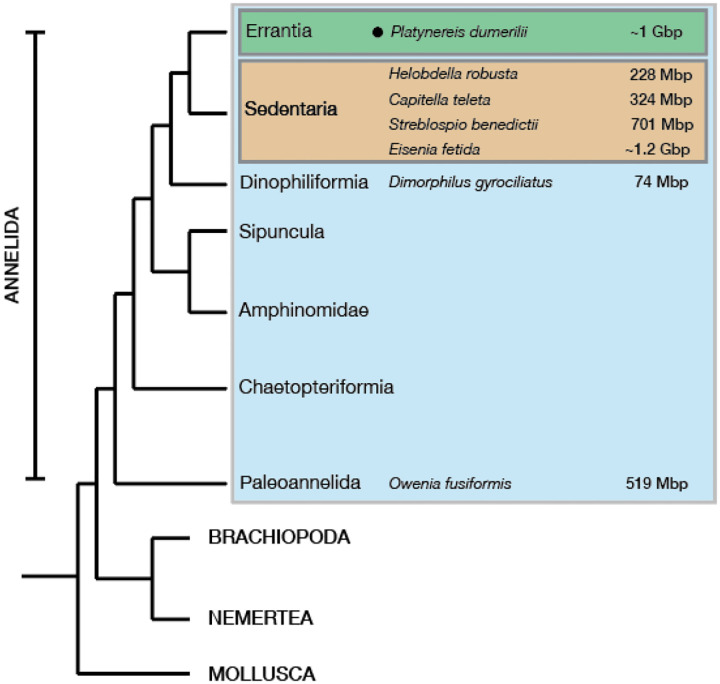
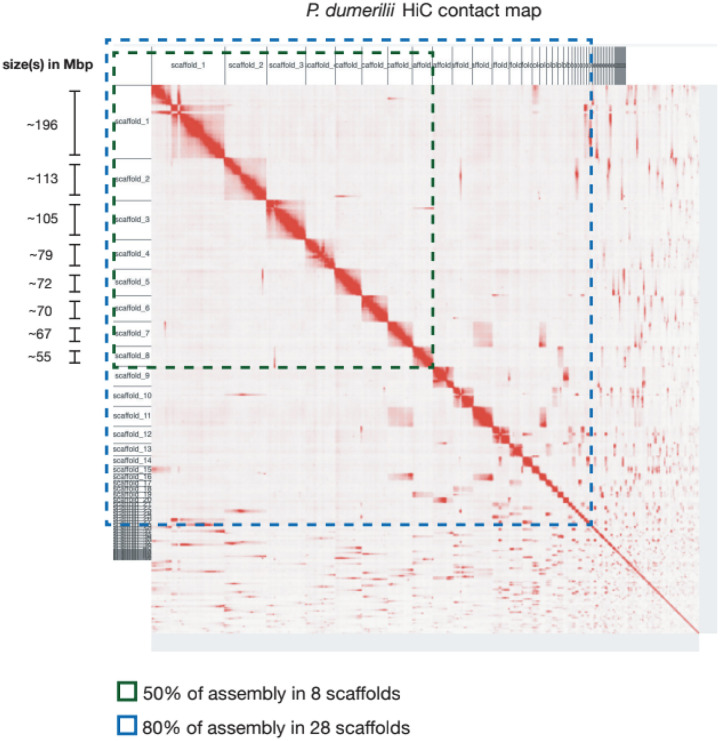
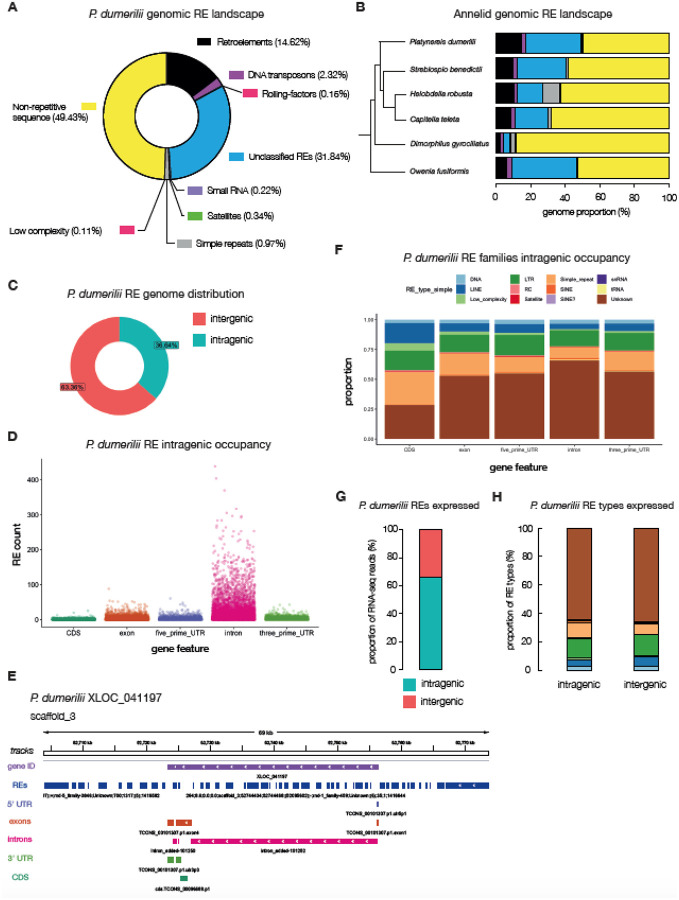
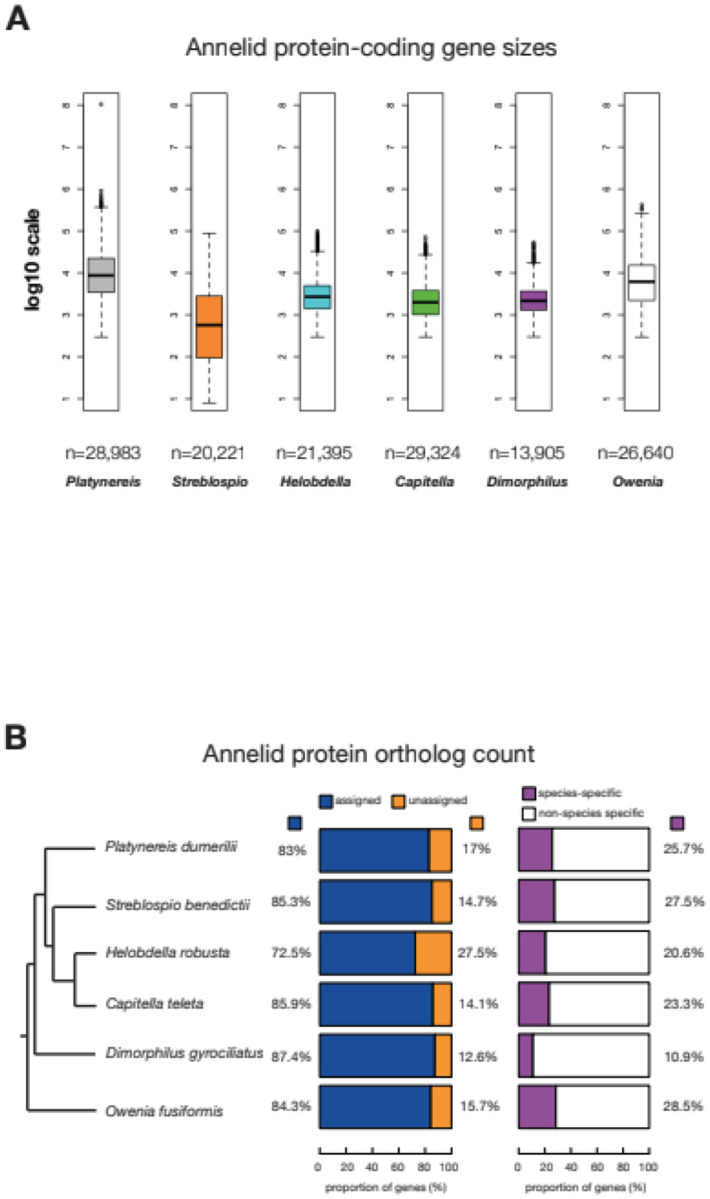
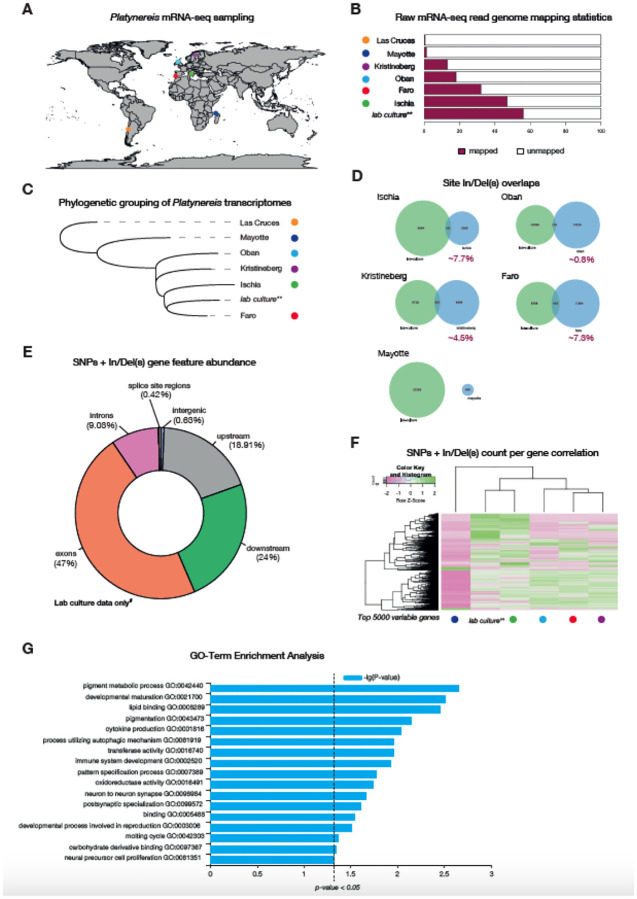
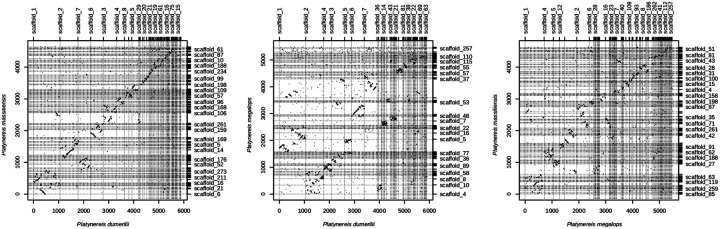
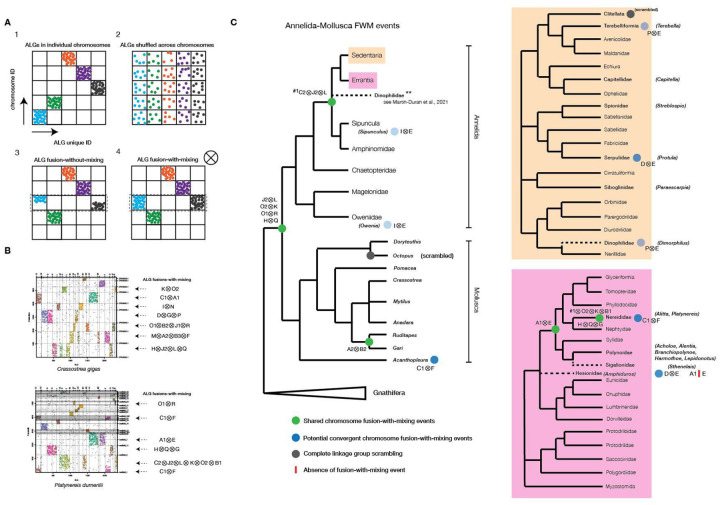
The marine annelid Platynereis dumerilii is a model organism used in many research areas including evolution and development, neurobiology, ecology and regeneration. Here we present the genomes of P. dumerilii (laboratory culture reference and a single individual assembly) and of the closely related P. massiliensis and P. megalops (single individual assembly) to facilitate comparative genomic approaches and help explore Platynereis biology. We used long-read sequencing technology and chromosomal-conformation capture along with extensive transcriptomic resources to obtain and annotate a draft genome assembly of ~1.47 Gbp for P. dumerilii, of which more than half represent repeat elements. We predict around 29,000 protein-coding genes, with relatively large intron sizes, over 38,000 non-coding genes, and 105 miRNA loci. We further explore the high genetic variation (~3% heterozygosity) within the Platynereis species complex. Gene ontology reveals the most variable loci to be associated with pigmentation, development and immunity. The current work sets the stage for further development of Platynereis genomic resources.
Keywords: Platynereis; Platynereis dumerilii; Spiralia; annelid; evo-devo; genome; model organism.
Figures

References
-
- Alves P. R., Halanych K. M., & Santos C. S. G. (2020). The phylogeny of Nereididae (Annelida) based on mitochondrial genomes. Zoologica Scripta, 49(3), 366–378. 10.1111/zsc.12413 - DOI
-
- Audouin J. V., & Milne Edwards H. (1833). [Part 3.] Classification des Annélides et description de celles qui habitent les côtes de la France. Annales Des Sciences Naturelles, Paris. https://biodiversitylibrary.org/page/6044727
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials