

YTHDC1 m6A-dependent and m6A-independent functions converge to preserve the DNA damage response
- PMID: 38951610
- PMCID: PMC11329685
- DOI: 10.1038/s44318-024-00153-x
YTHDC1 m6A-dependent and m6A-independent functions converge to preserve the DNA damage response
Abstract
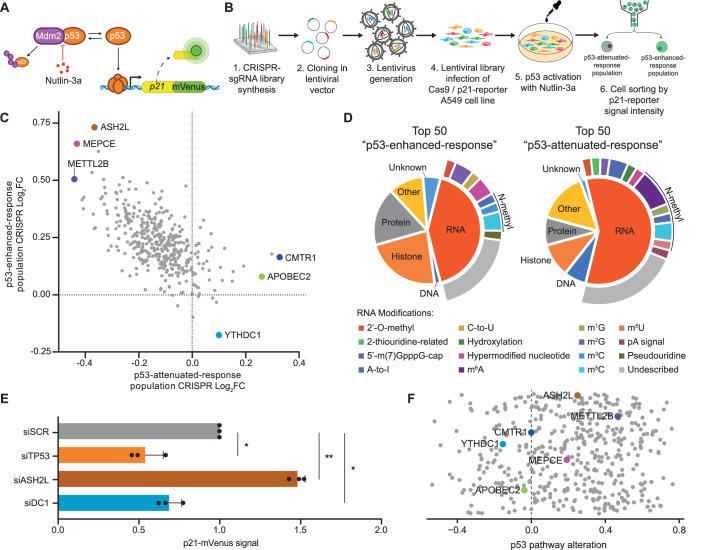
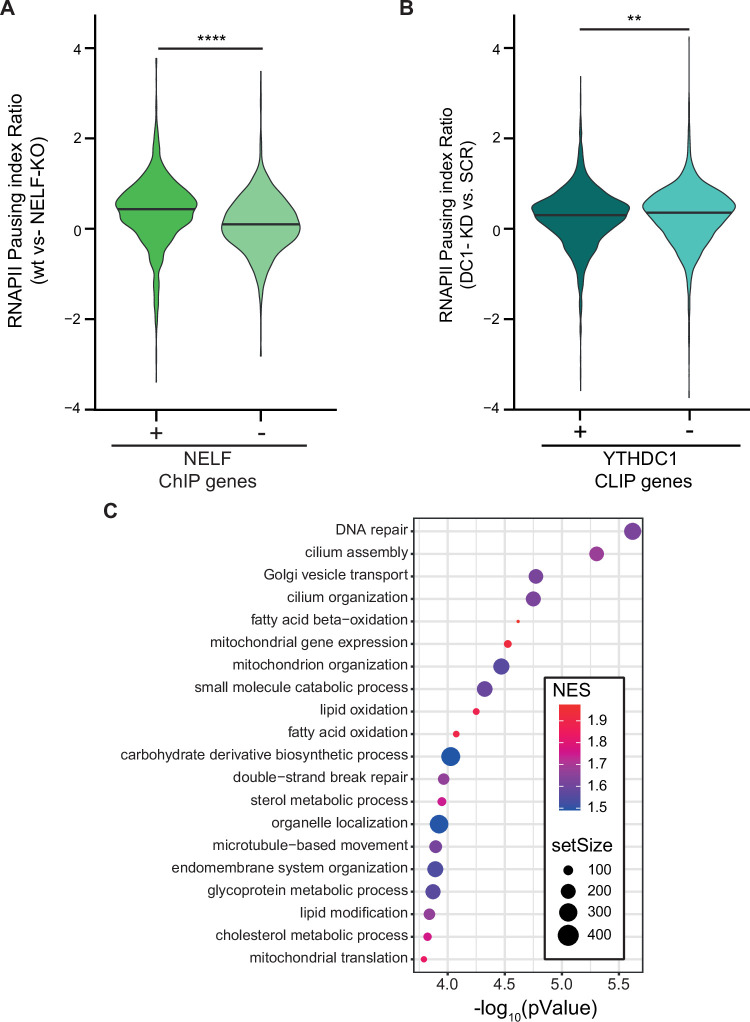
Cells have evolved a robust and highly regulated DNA damage response to preserve their genomic integrity. Although increasing evidence highlights the relevance of RNA regulation, our understanding of its impact on a fully efficient DNA damage response remains limited. Here, through a targeted CRISPR-knockout screen, we identify RNA-binding proteins and modifiers that participate in the p53 response. Among the top hits, we find the m6A reader YTHDC1 as a master regulator of p53 expression. YTHDC1 binds to the transcription start sites of TP53 and other genes involved in the DNA damage response, promoting their transcriptional elongation. YTHDC1 deficiency also causes the retention of introns and therefore aberrant protein production of key DNA damage factors. While YTHDC1-mediated intron retention requires m6A, TP53 transcriptional pause-release is promoted by YTHDC1 independently of m6A. Depletion of YTHDC1 causes genomic instability and aberrant cancer cell proliferation mediated by genes regulated by YTHDC1. Our results uncover YTHDC1 as an orchestrator of the DNA damage response through distinct mechanisms of co-transcriptional mRNA regulation.
Keywords: DDR; RNAPII Pausing; Splicing; YTHDC1; p53.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures









Similar articles
-
YTHDC1 is a therapeutic target for B-cell acute lymphoblastic leukemia by attenuating DNA damage response through the KMT2C-H3K4me1/me3 epigenetic axis.Leukemia. 2025 Feb;39(2):308-322. doi: 10.1038/s41375-024-02451-z. Epub 2024 Nov 5. Leukemia. 2025. PMID: 39501105
-
YTHDC1 cooperates with the THO complex to prevent RNA-damage-induced DNA breaks.Mol Cell. 2025 Mar 20;85(6):1085-1100.e9. doi: 10.1016/j.molcel.2025.02.003. Epub 2025 Mar 3. Mol Cell. 2025. PMID: 40037355
-
Nuclear Aurora kinase A switches m6A reader YTHDC1 to enhance an oncogenic RNA splicing of tumor suppressor RBM4.Signal Transduct Target Ther. 2022 Apr 1;7(1):97. doi: 10.1038/s41392-022-00905-3. Signal Transduct Target Ther. 2022. PMID: 35361747 Free PMC article.
-
m(6)A: Signaling for mRNA splicing.RNA Biol. 2016 Sep;13(9):756-9. doi: 10.1080/15476286.2016.1201628. Epub 2016 Jun 28. RNA Biol. 2016. PMID: 27351695 Free PMC article. Review.
-
The multifaceted effects of YTHDC1-mediated nuclear m6A recognition.Trends Genet. 2022 Apr;38(4):325-332. doi: 10.1016/j.tig.2021.11.005. Epub 2021 Dec 14. Trends Genet. 2022. PMID: 34920906 Review.
Cited by
-
DUX4-induced HSATII RNA accumulation drives protein aggregation impacting RNA processing pathways.bioRxiv [Preprint]. 2025 Jan 22:2024.12.17.628988. doi: 10.1101/2024.12.17.628988. bioRxiv. 2025. PMID: 39764024 Free PMC article. Preprint.
-
Decoding the molecular symphony: interactions between the m6A and p53 signaling pathways in cancer.NAR Cancer. 2024 Sep 26;6(3):zcae037. doi: 10.1093/narcan/zcae037. eCollection 2024 Sep. NAR Cancer. 2024. PMID: 39329012 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
