

Asymmetric apical domain states of mitochondrial Hsp60 coordinate substrate engagement and chaperonin assembly
- PMID: 38951622
- PMCID: PMC11638070
- DOI: 10.1038/s41594-024-01352-0
Asymmetric apical domain states of mitochondrial Hsp60 coordinate substrate engagement and chaperonin assembly
Abstract
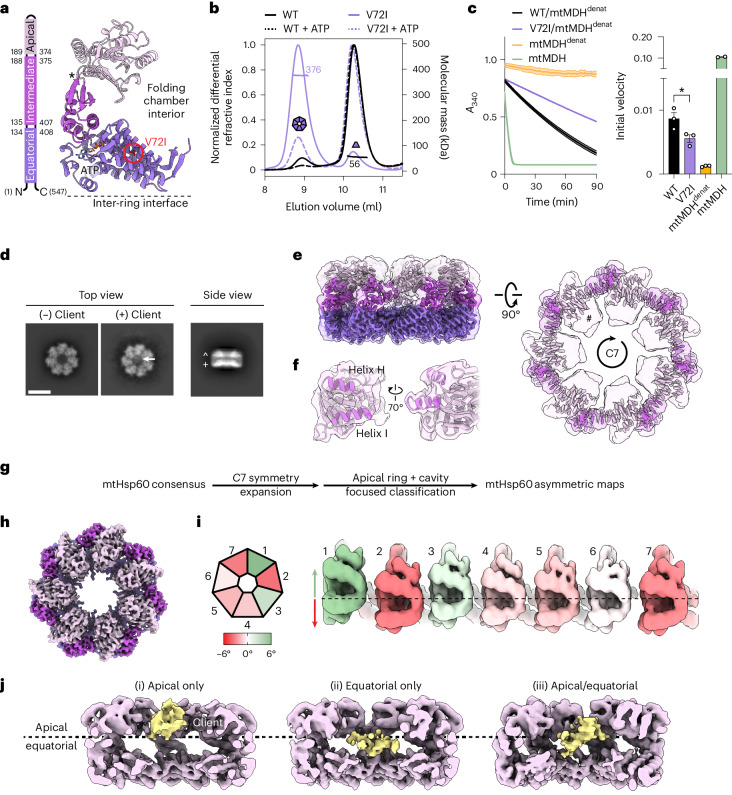
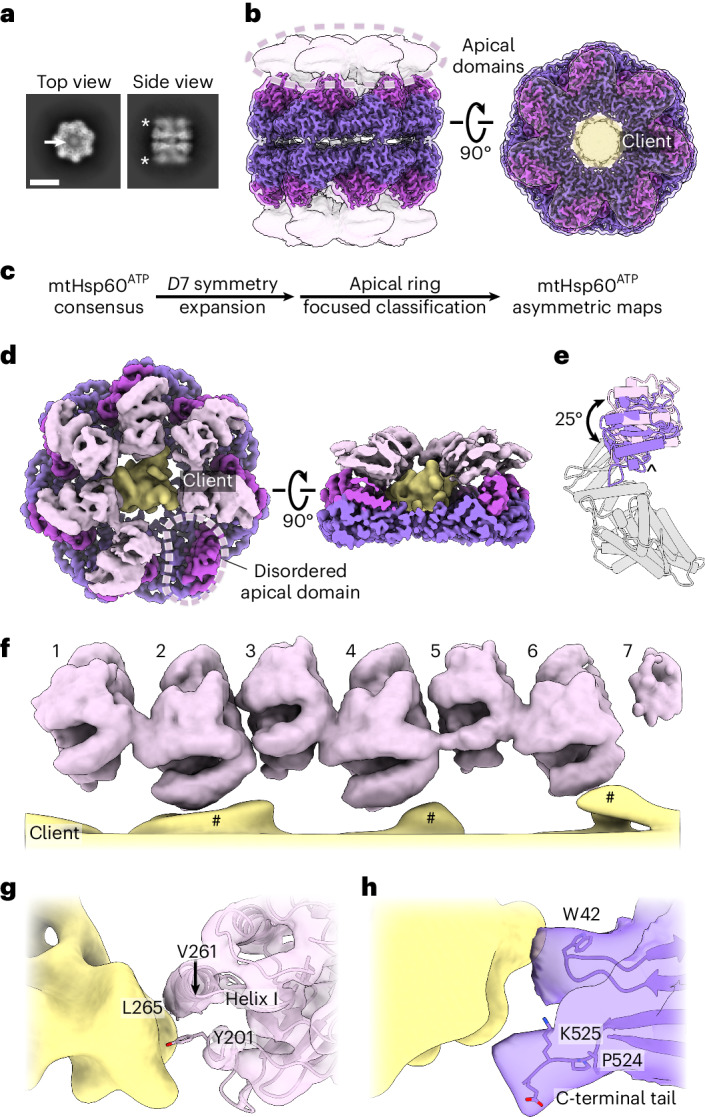
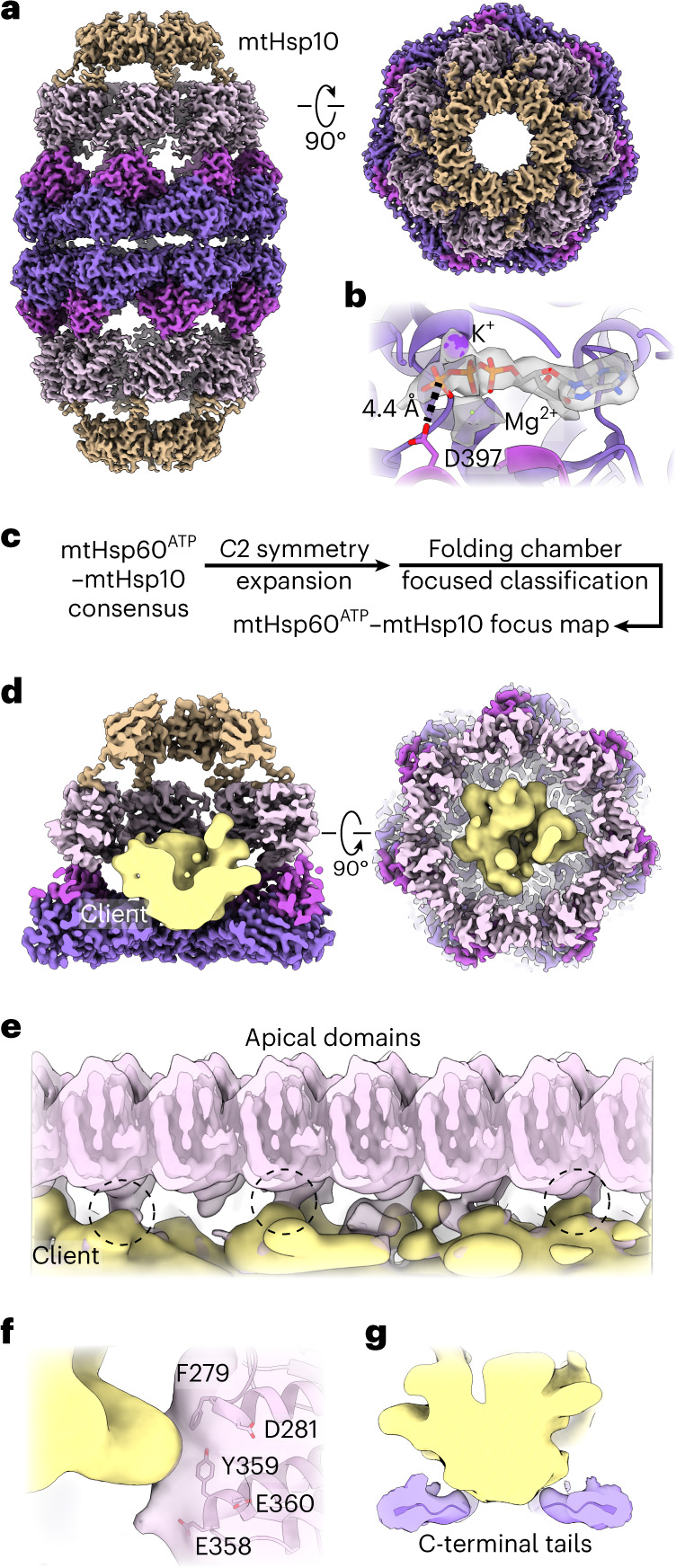
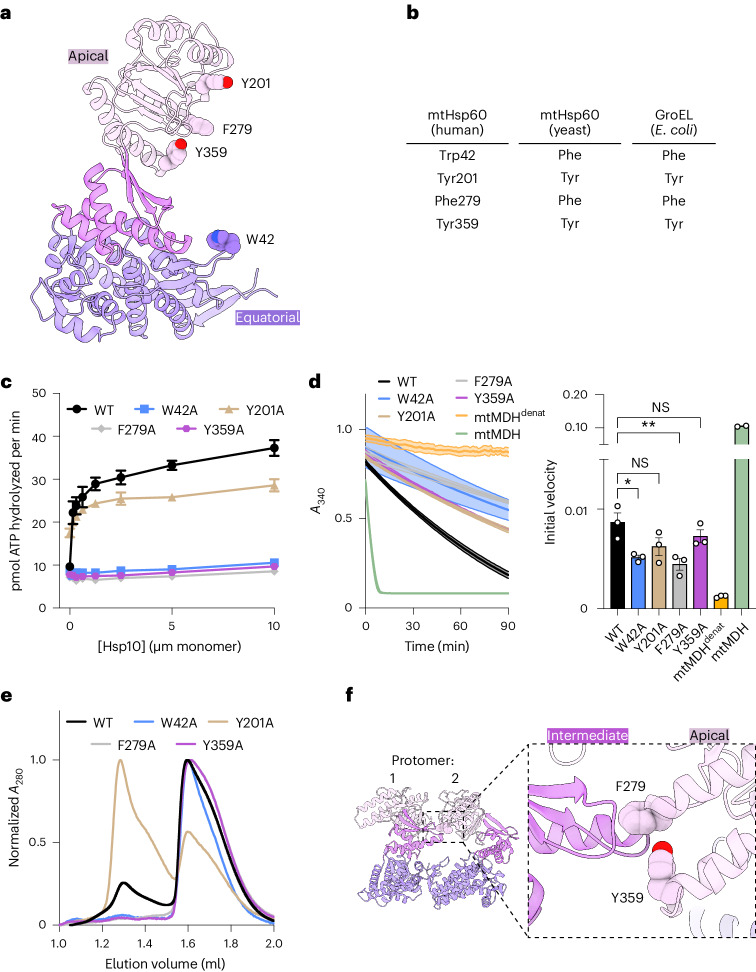
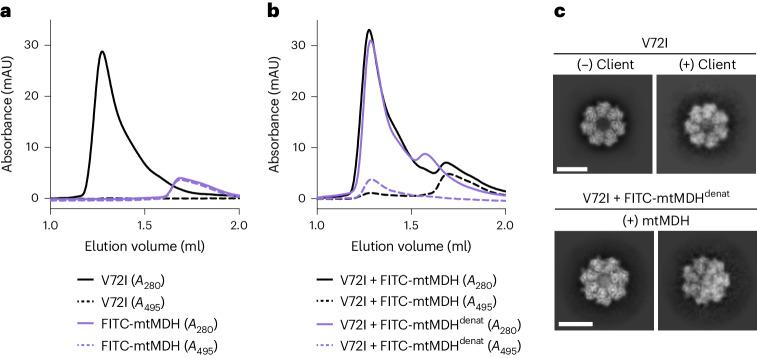
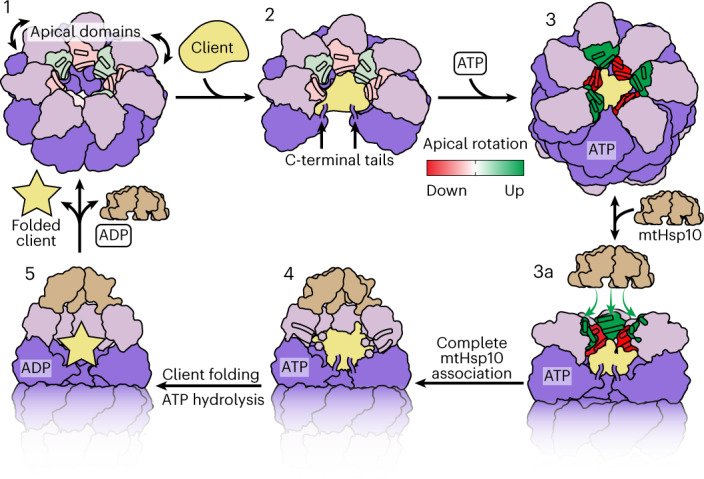
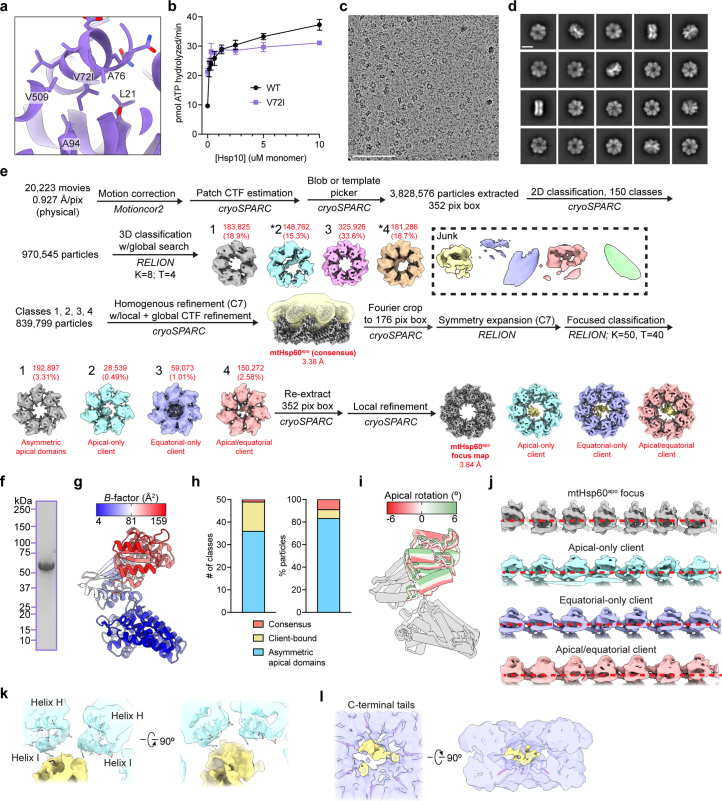
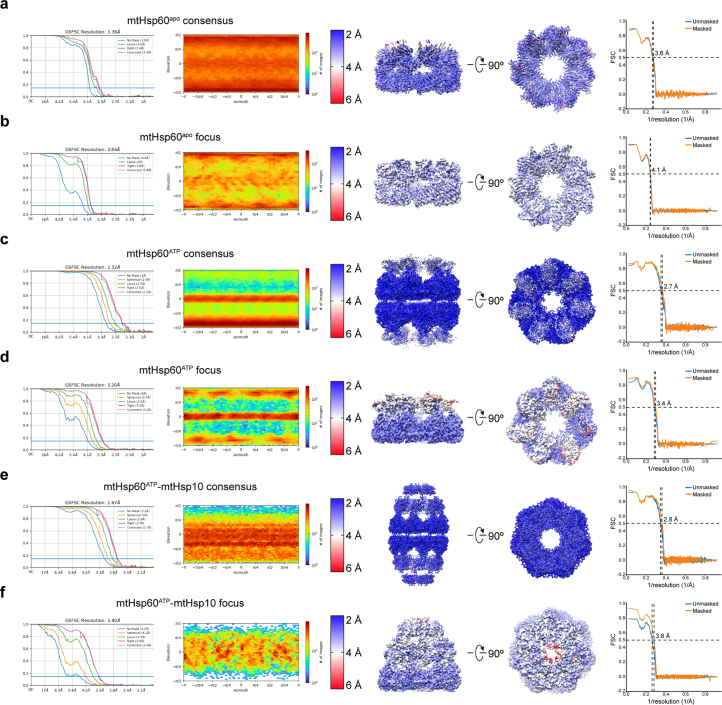
The mitochondrial chaperonin, mitochondrial heat shock protein 60 (mtHsp60), promotes the folding of newly imported and transiently misfolded proteins in the mitochondrial matrix, assisted by its co-chaperone mtHsp10. Despite its essential role in mitochondrial proteostasis, structural insights into how this chaperonin progresses through its ATP-dependent client folding cycle are not clear. Here, we determined cryo-EM structures of a hyperstable disease-associated human mtHsp60 mutant, V72I. Client density is identified in three distinct states, revealing interactions with the mtHsp60 apical domains and C termini that coordinate client positioning in the folding chamber. We further identify an asymmetric arrangement of the apical domains in the ATP state, in which an alternating up/down configuration positions interaction surfaces for simultaneous recruitment of mtHsp10 and client retention. Client is then fully encapsulated in mtHsp60-10, revealing prominent contacts at two discrete sites that potentially support maturation. These results identify distinct roles for the apical domains in coordinating client capture and progression through the chaperone cycle, supporting a conserved mechanism of group I chaperonin function.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures


Update of
-
Asymmetric apical domain states of mitochondrial Hsp60 coordinate substrate engagement and chaperonin assembly.bioRxiv [Preprint]. 2023 May 15:2023.05.15.540872. doi: 10.1101/2023.05.15.540872. bioRxiv. 2023. Update in: Nat Struct Mol Biol. 2024 Dec;31(12):1848-1858. doi: 10.1038/s41594-024-01352-0. PMID: 37293102 Free PMC article. Updated. Preprint.
References
-
- Balchin, D., Hayer-Hartl, M. & Hartl, F. U. In vivo aspects of protein folding and quality control. Science353, aac4354 (2016). - PubMed
-
- Horwich, A. L. & Fenton, W. A. Chaperonin-assisted protein folding: a chronologue. Q. Rev. Biophys.53, e4 (2020). - PubMed
-
- Yébenes, H., Mesa, P., Muñoz, I. G., Montoya, G. & Valpuesta, J. M. Chaperonins: two rings for folding. Trends Biochem. Sci.36, 424–432 (2011). - PubMed
-
- Hayer-Hartl, M., Bracher, A. & Hartl, F. U. The GroEL–GroES chaperonin machine: a nano-cage for protein folding. Trends Biochem. Sci.41, 62–76 (2016). - PubMed
-
- Fenton, W. A., Kashi, Y., Furtak, K. & Horwich, A. L. Residues in chaperonin GroEL required for polypeptide binding and release. Nature371, 614–619 (1994). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
