

Mapping spatially resolved transcriptomes in human and mouse pulmonary fibrosis
- PMID: 38951642
- PMCID: PMC11319205
- DOI: 10.1038/s41588-024-01819-2
Mapping spatially resolved transcriptomes in human and mouse pulmonary fibrosis
Abstract
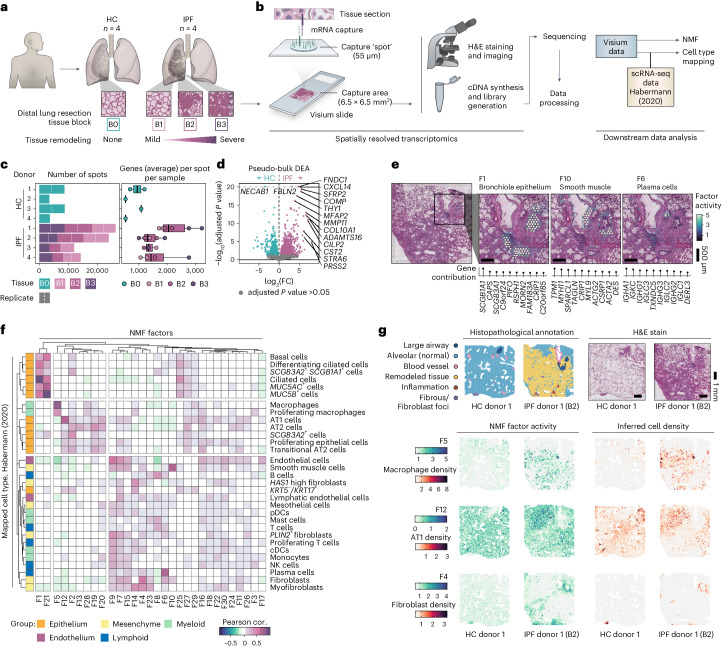
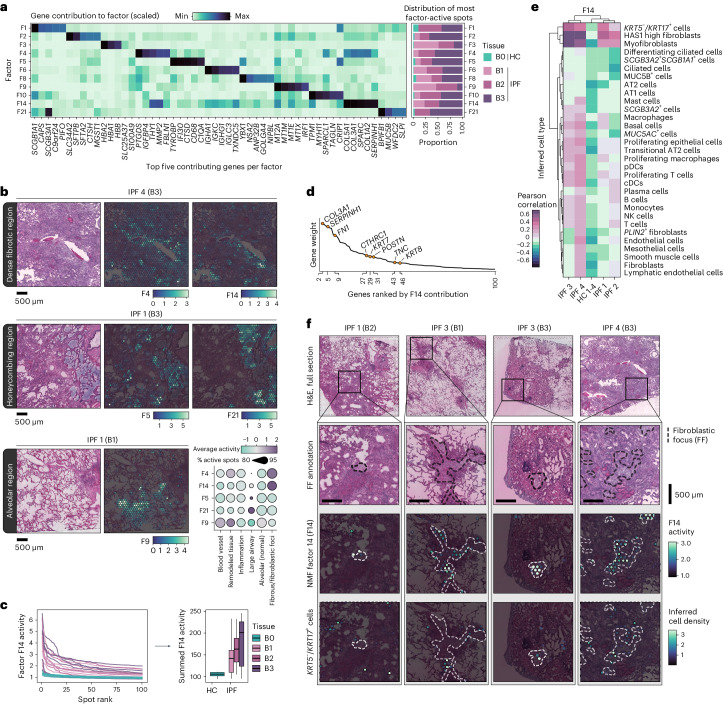
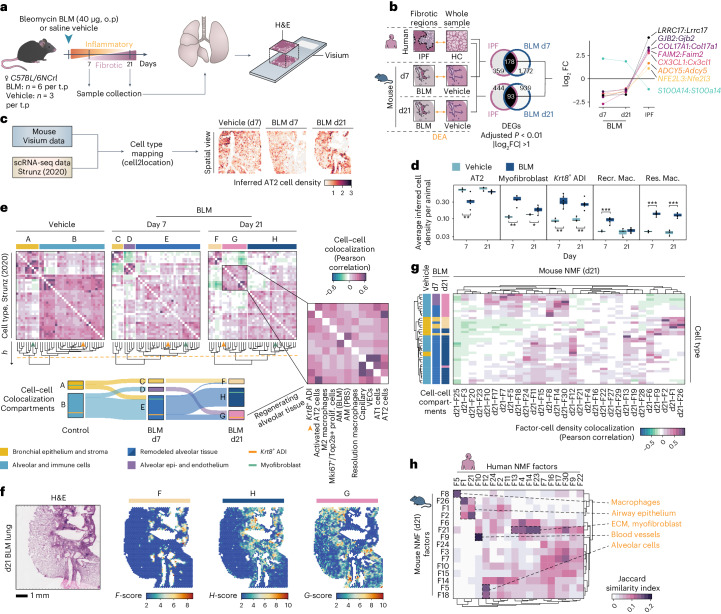
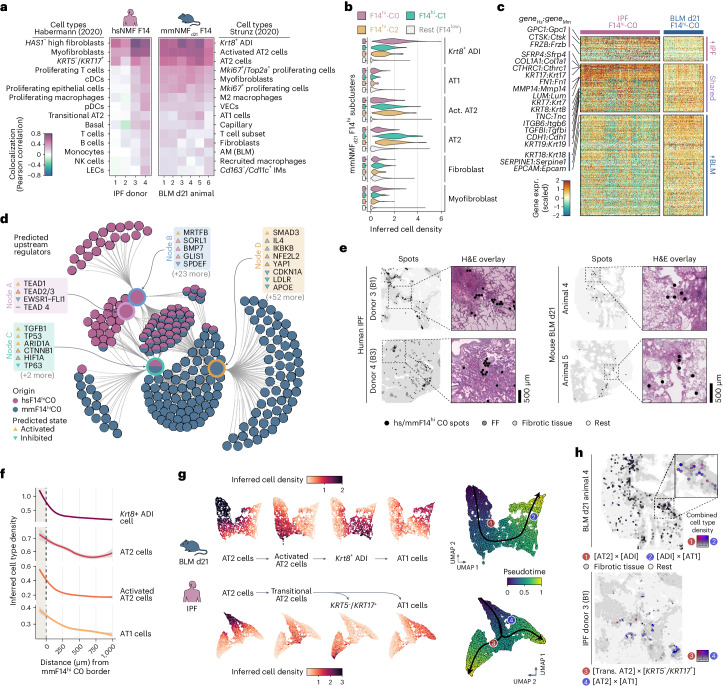
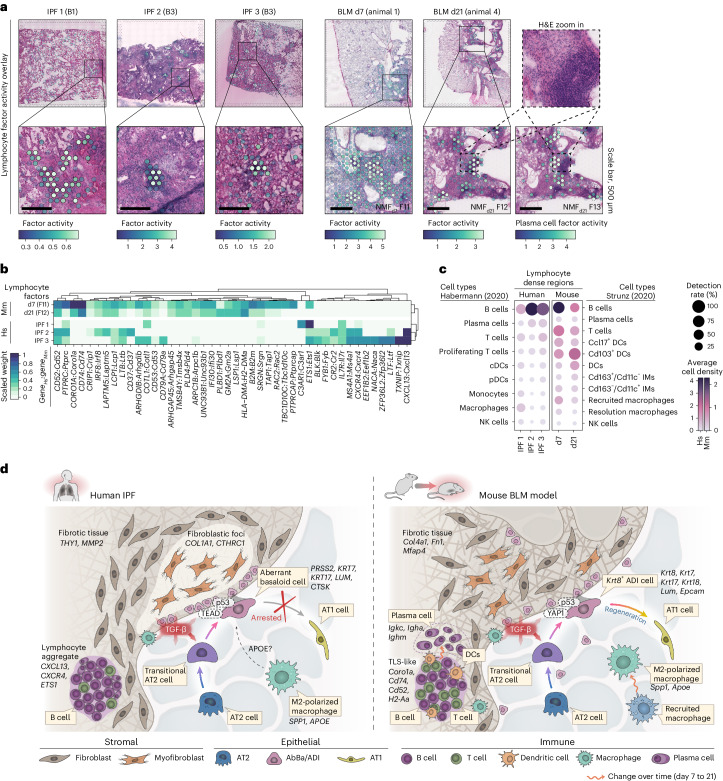
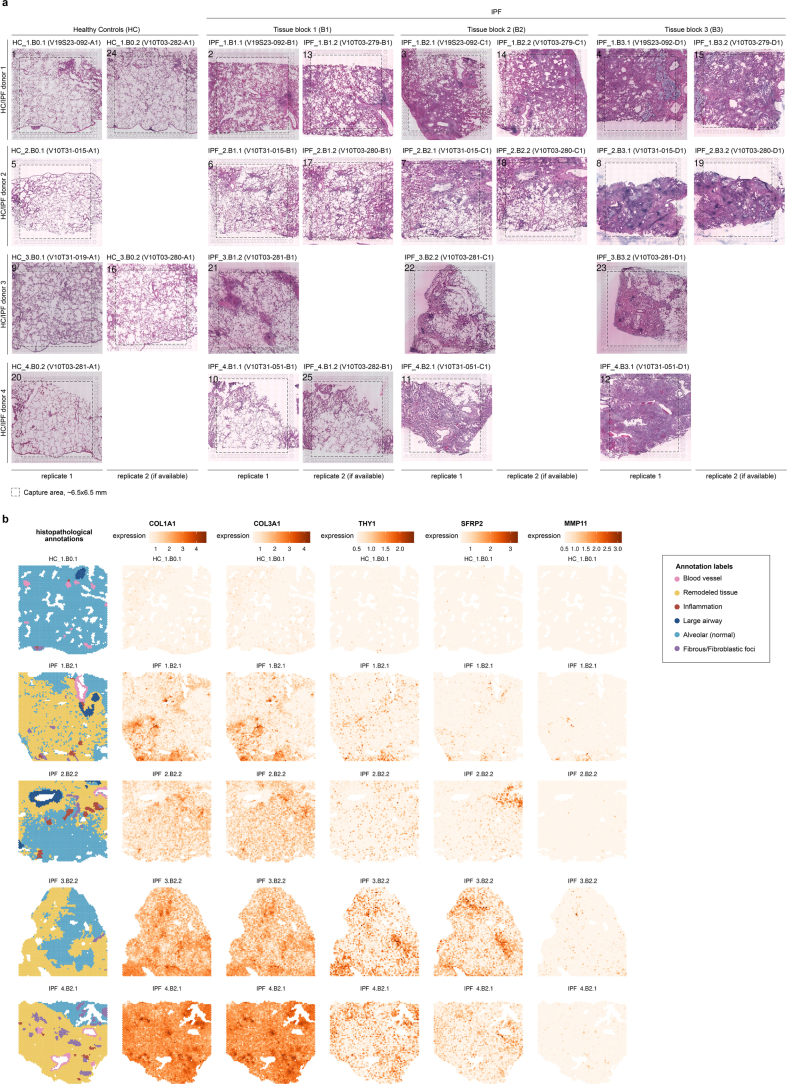
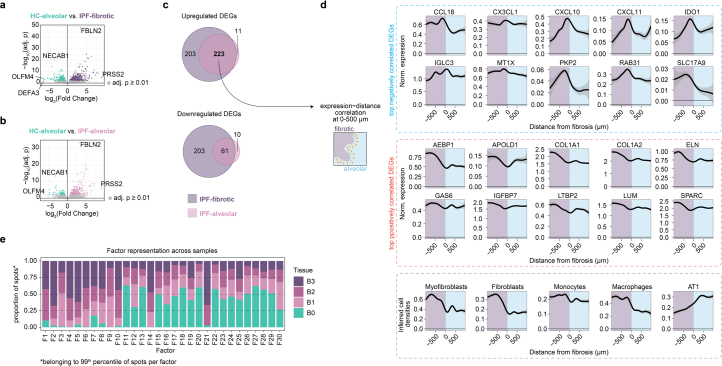
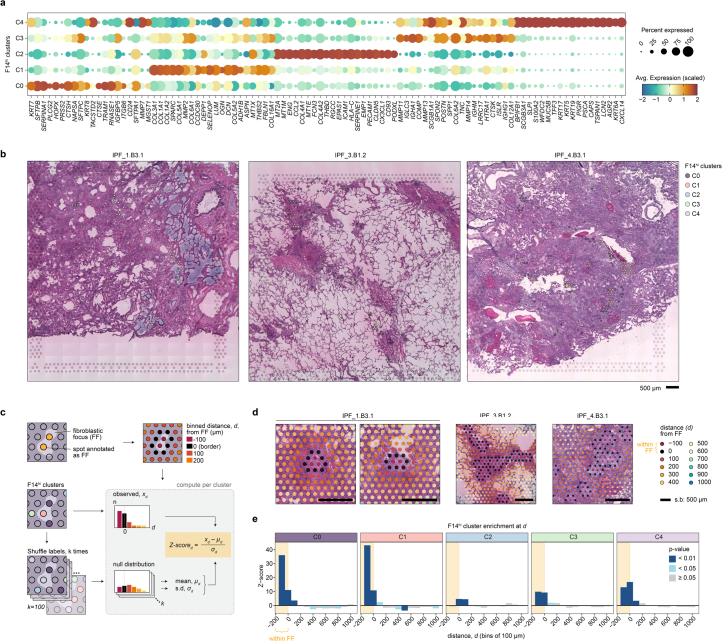
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with poor prognosis and limited treatment options. Efforts to identify effective treatments are thwarted by limited understanding of IPF pathogenesis and poor translatability of available preclinical models. Here we generated spatially resolved transcriptome maps of human IPF (n = 4) and bleomycin-induced mouse pulmonary fibrosis (n = 6) to address these limitations. We uncovered distinct fibrotic niches in the IPF lung, characterized by aberrant alveolar epithelial cells in a microenvironment dominated by transforming growth factor beta signaling alongside predicted regulators, such as TP53 and APOE. We also identified a clear divergence between the arrested alveolar regeneration in the IPF fibrotic niches and the active tissue repair in the acutely fibrotic mouse lung. Our study offers in-depth insights into the IPF transcriptional landscape and proposes alveolar regeneration as a promising therapeutic strategy for IPF.
© 2024. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: P.L.S. and J. Lundeberg are scientific consultants to 10x Genomics. The remaining authors (L.F., M.O.L., M.H., V.P., L.S., B.P.K., A.C., S.O., T.V., A.B., J. Lindgren, G.B., S.J., A.O., M.S. and J.H.) are employees and/or stockholders of AstraZeneca.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
