

Direct neuronal reprogramming of mouse astrocytes is associated with multiscale epigenome remodeling and requires Yy1
- PMID: 38956165
- PMCID: PMC11239498
- DOI: 10.1038/s41593-024-01677-5
Direct neuronal reprogramming of mouse astrocytes is associated with multiscale epigenome remodeling and requires Yy1
Abstract
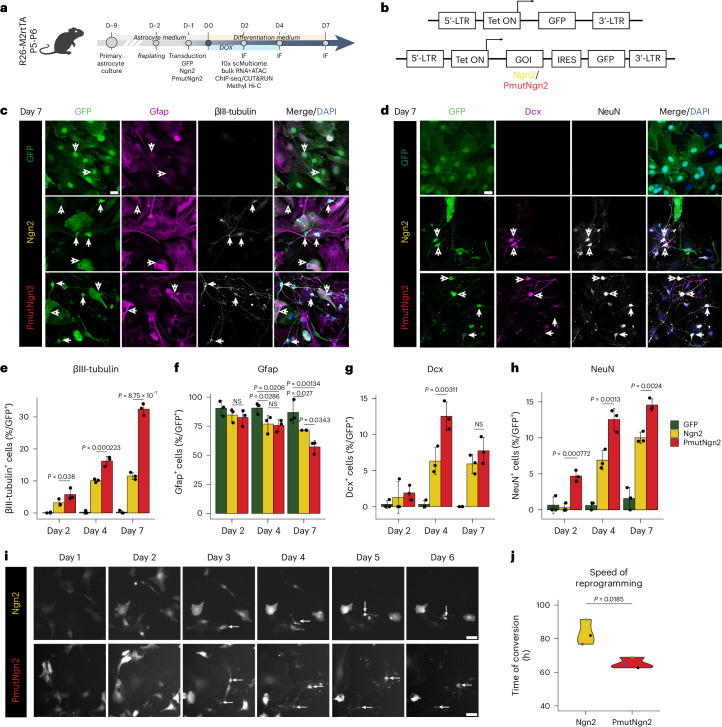
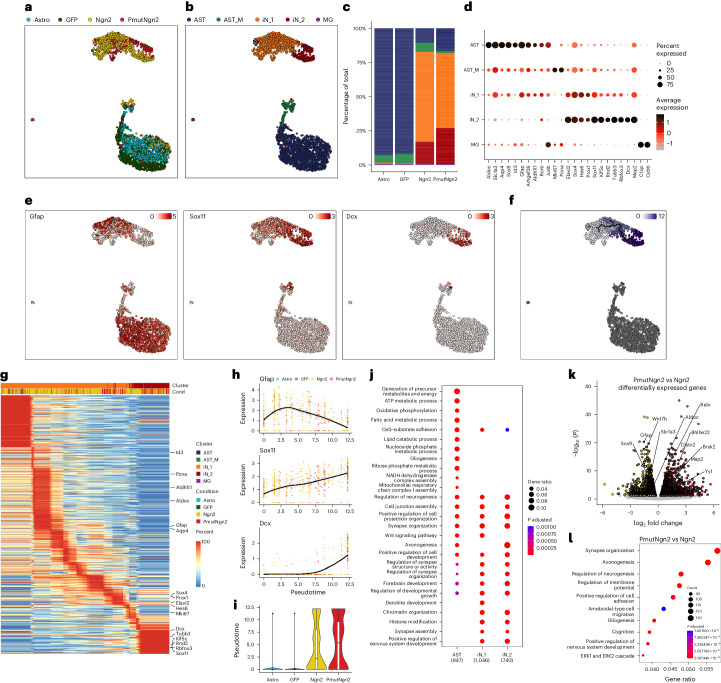
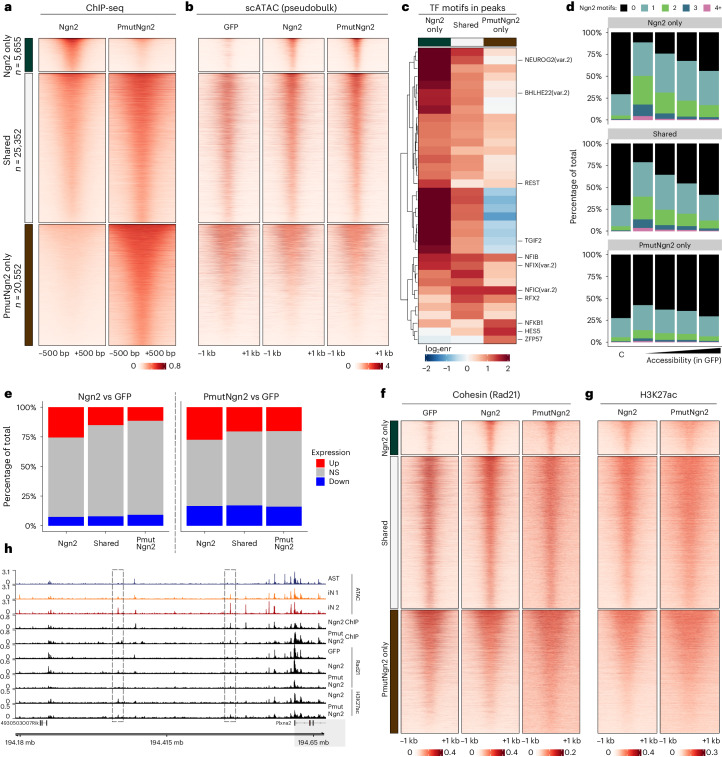
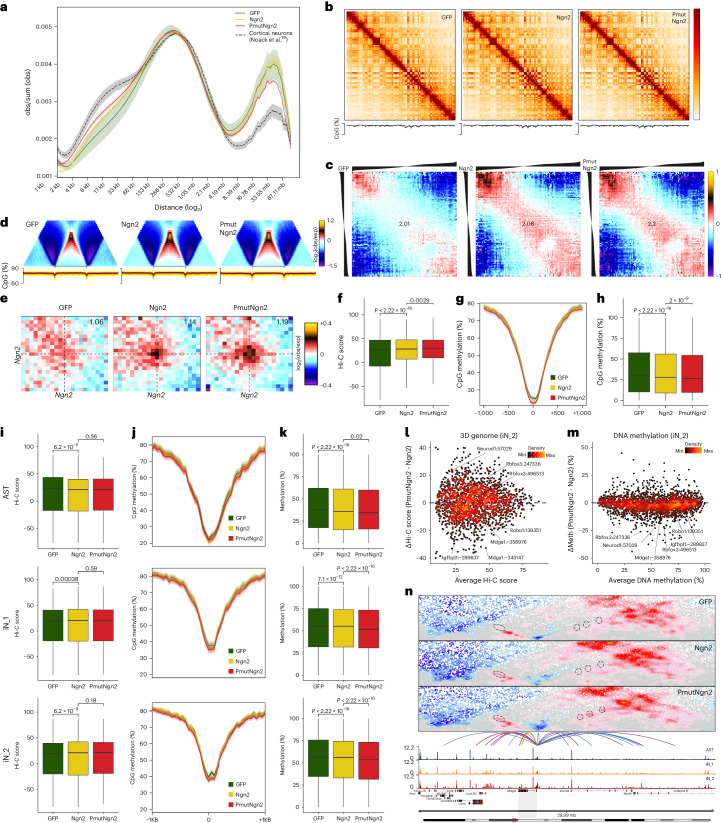
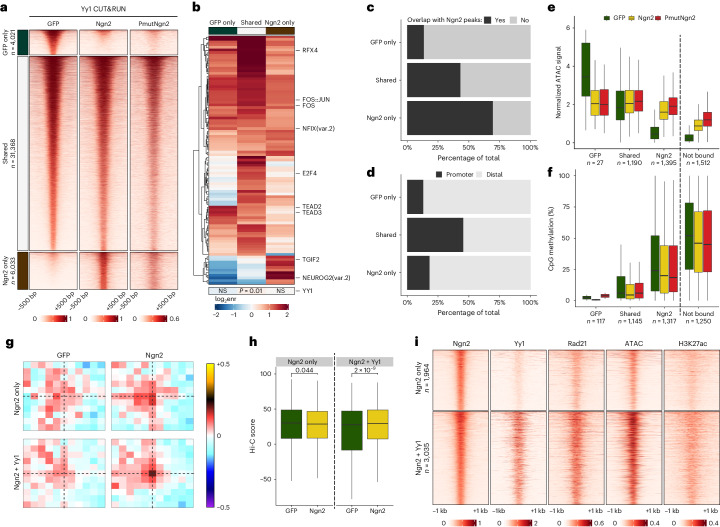
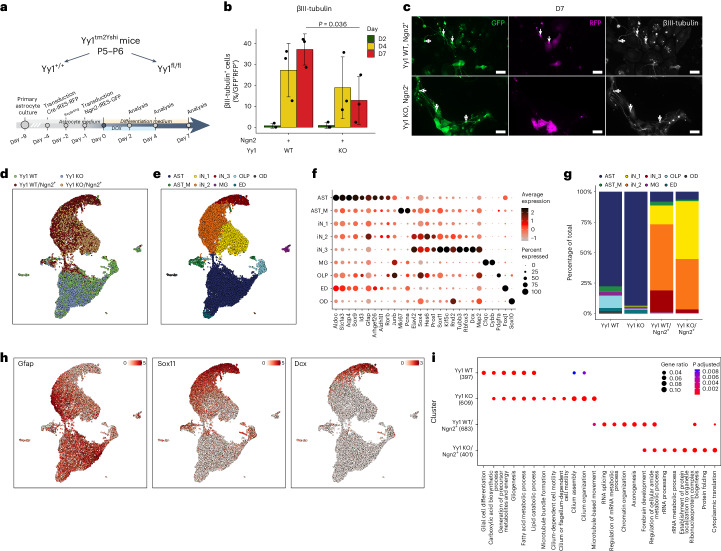
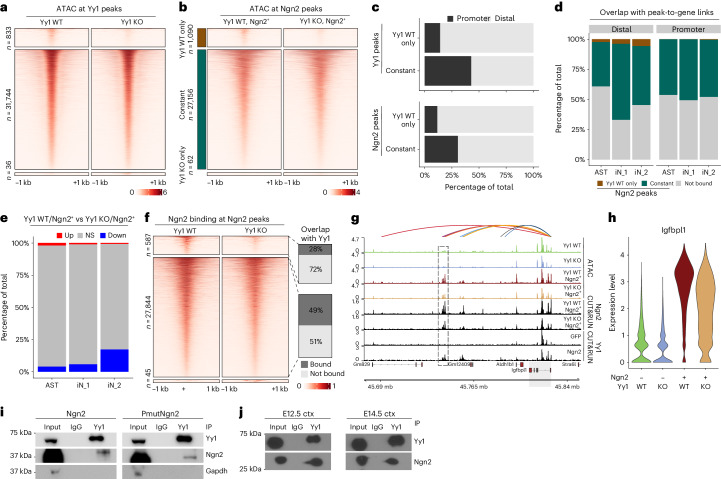
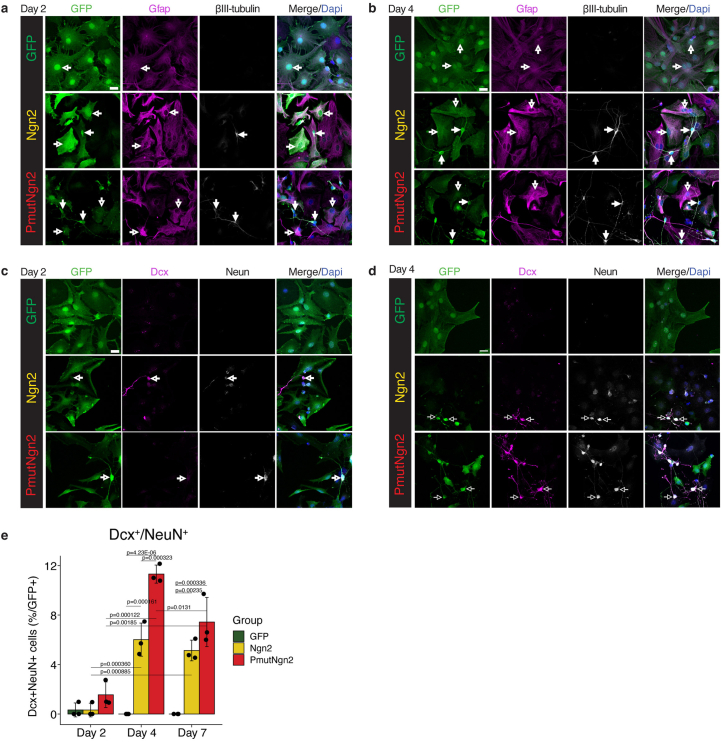
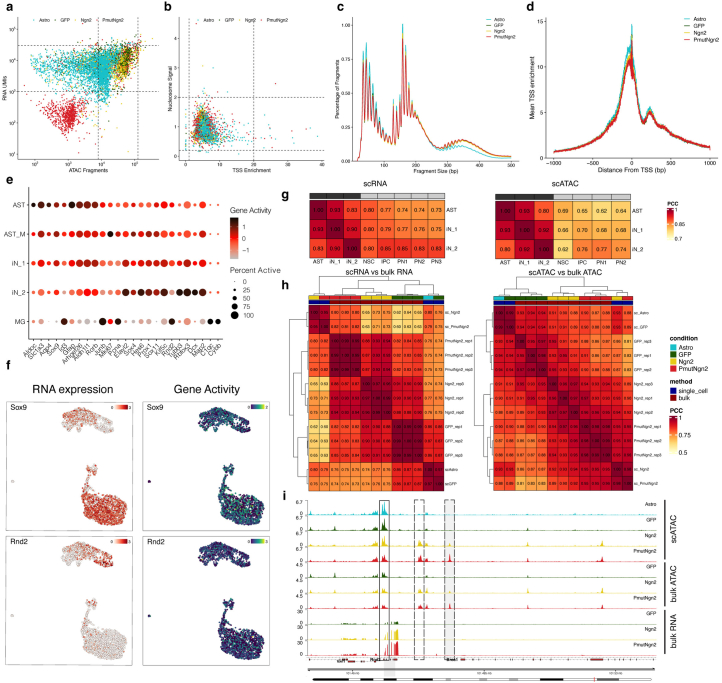
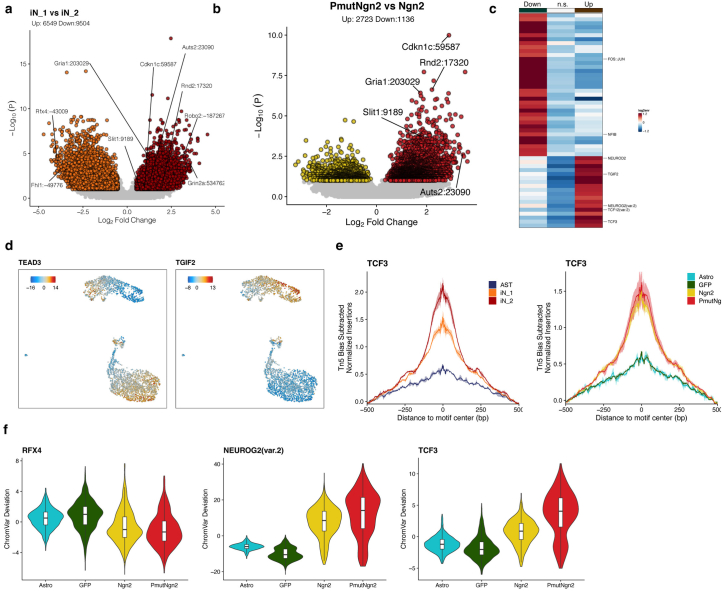
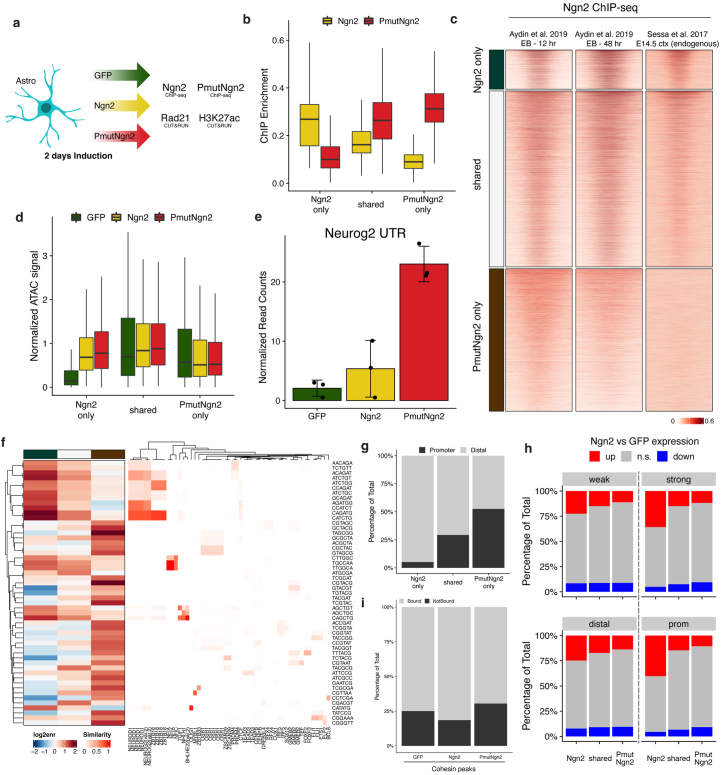
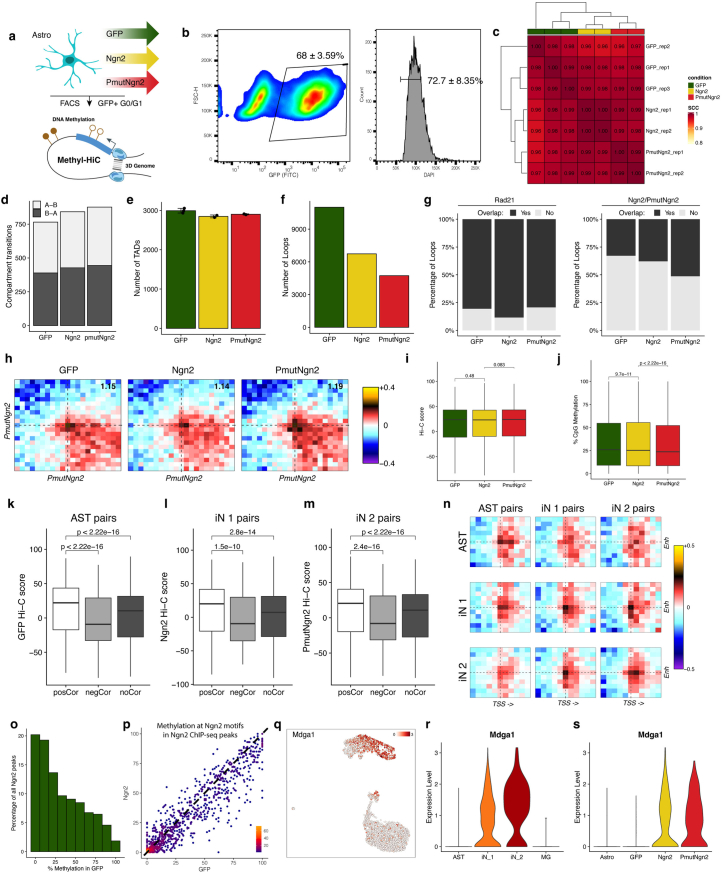
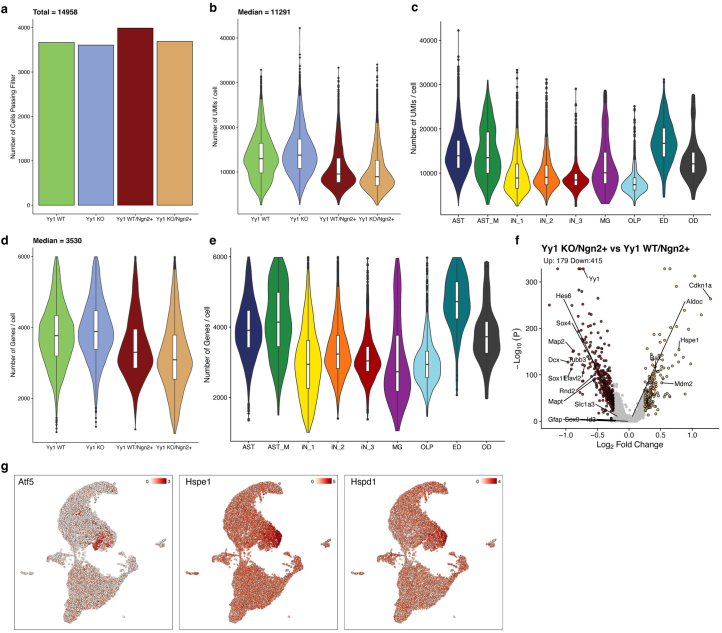
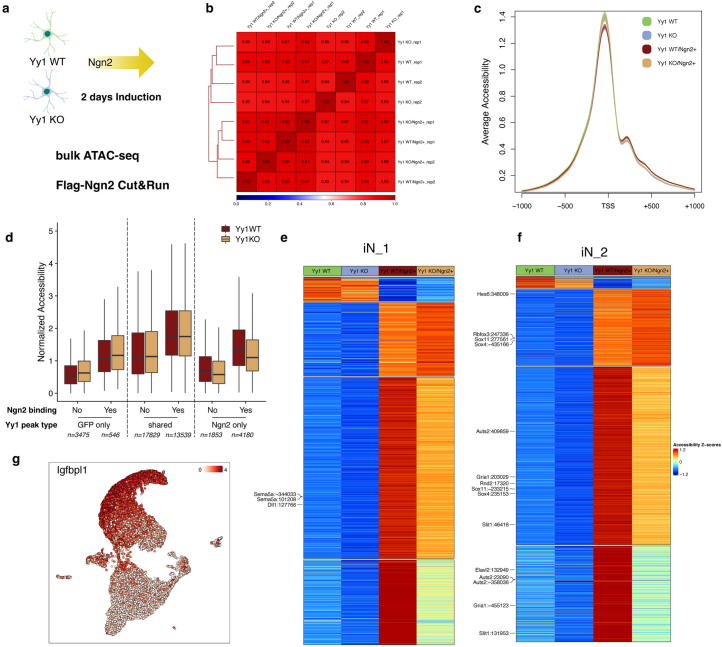
Direct neuronal reprogramming is a promising approach to regenerate neurons from local glial cells. However, mechanisms of epigenome remodeling and co-factors facilitating this process are unclear. In this study, we combined single-cell multiomics with genome-wide profiling of three-dimensional nuclear architecture and DNA methylation in mouse astrocyte-to-neuron reprogramming mediated by Neurogenin2 (Ngn2) and its phosphorylation-resistant form (PmutNgn2), respectively. We show that Ngn2 drives multilayered chromatin remodeling at dynamic enhancer-gene interaction sites. PmutNgn2 leads to higher reprogramming efficiency and enhances epigenetic remodeling associated with neuronal maturation. However, the differences in binding sites or downstream gene activation cannot fully explain this effect. Instead, we identified Yy1, a transcriptional co-factor recruited by direct interaction with Ngn2 to its target sites. Upon deletion of Yy1, activation of neuronal enhancers, genes and ultimately reprogramming are impaired without affecting Ngn2 binding. Thus, our work highlights the key role of interactors of proneural factors in direct neuronal reprogramming.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Barker RA, Gotz M, Parmar M. New approaches for brain repair—from rescue to reprogramming. Nature. 2018;557:329–334. - PubMed
-
- Heins N, et al. Glial cells generate neurons: the role of the transcription factor Pax6. Nat. Neurosci. 2002;5:308–315. - PubMed
-
- Gascón, S. et al. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell18, 396–409 (2016). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
