

Molecular taxonomy of myelodysplastic syndromes and its clinical implications
- PMID: 38958467
- PMCID: PMC11487646
- DOI: 10.1182/blood.2023023727
Molecular taxonomy of myelodysplastic syndromes and its clinical implications
Abstract
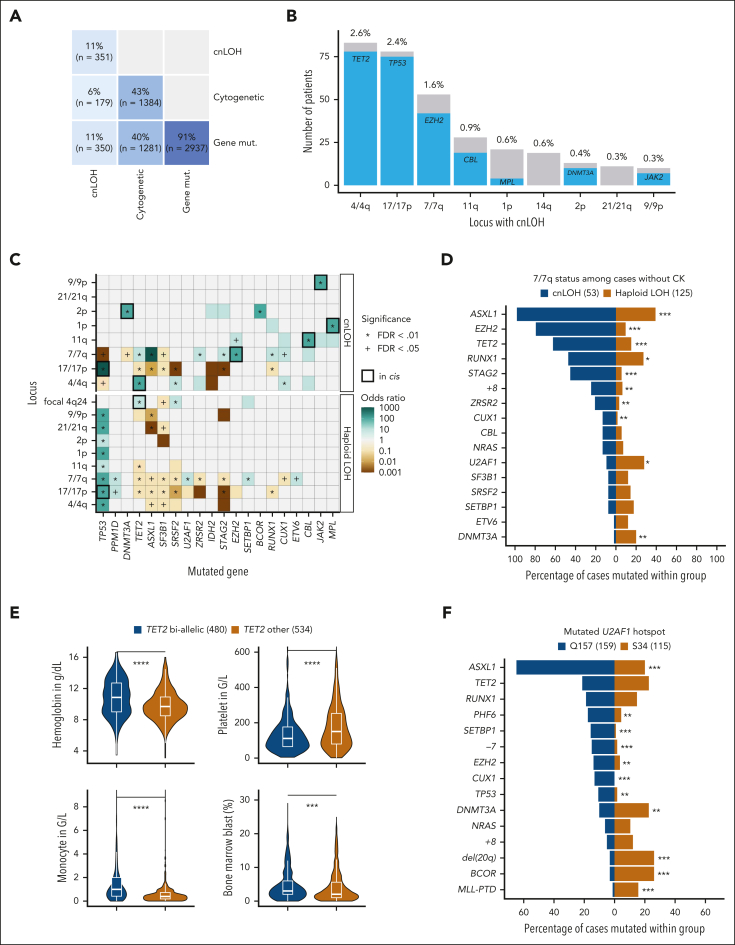
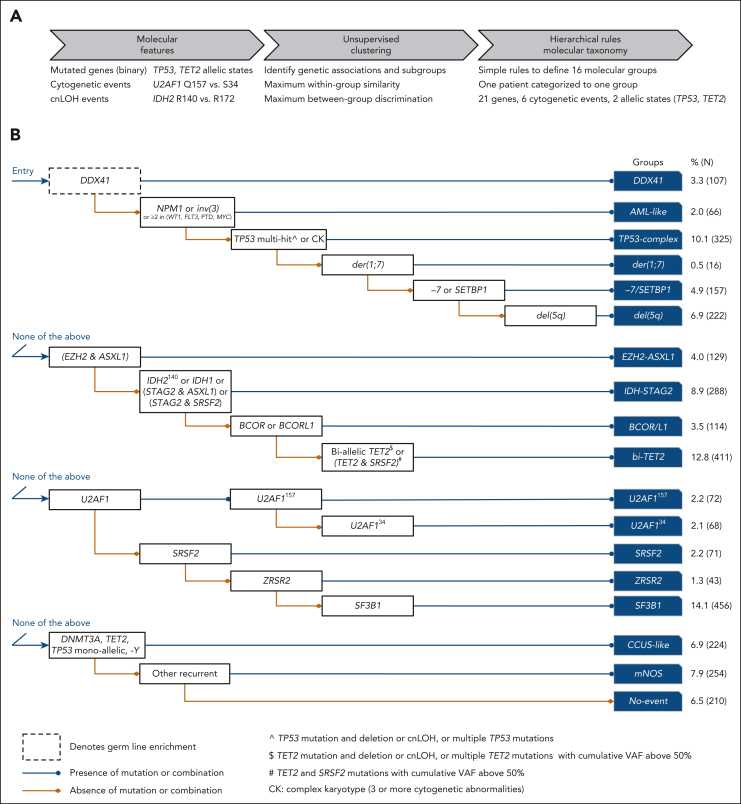
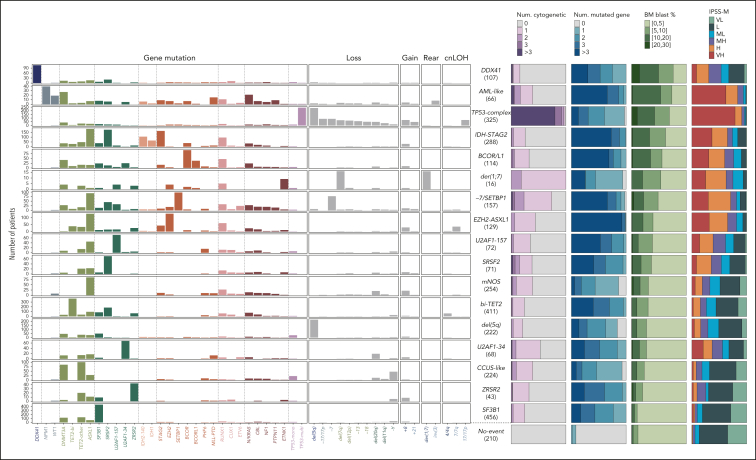
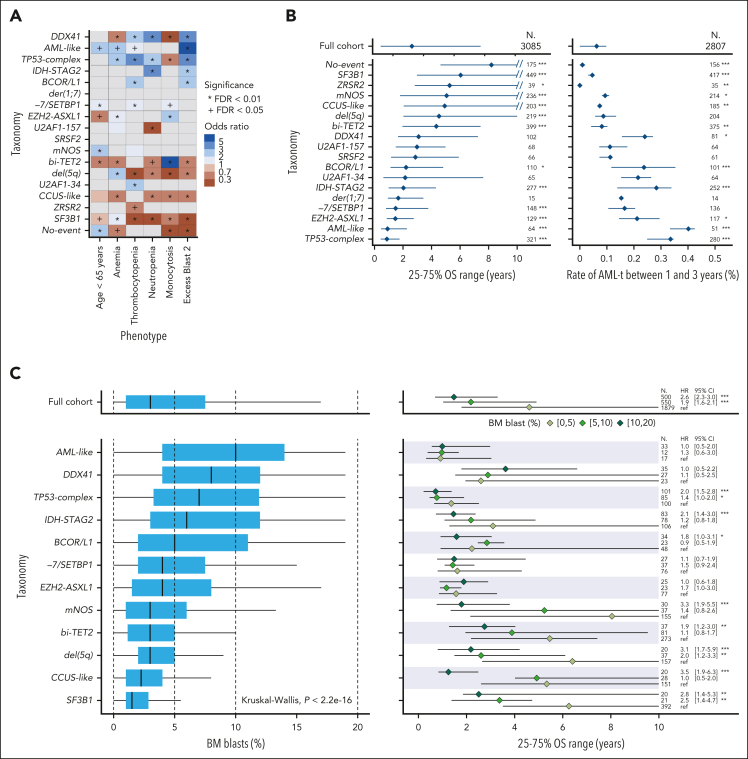
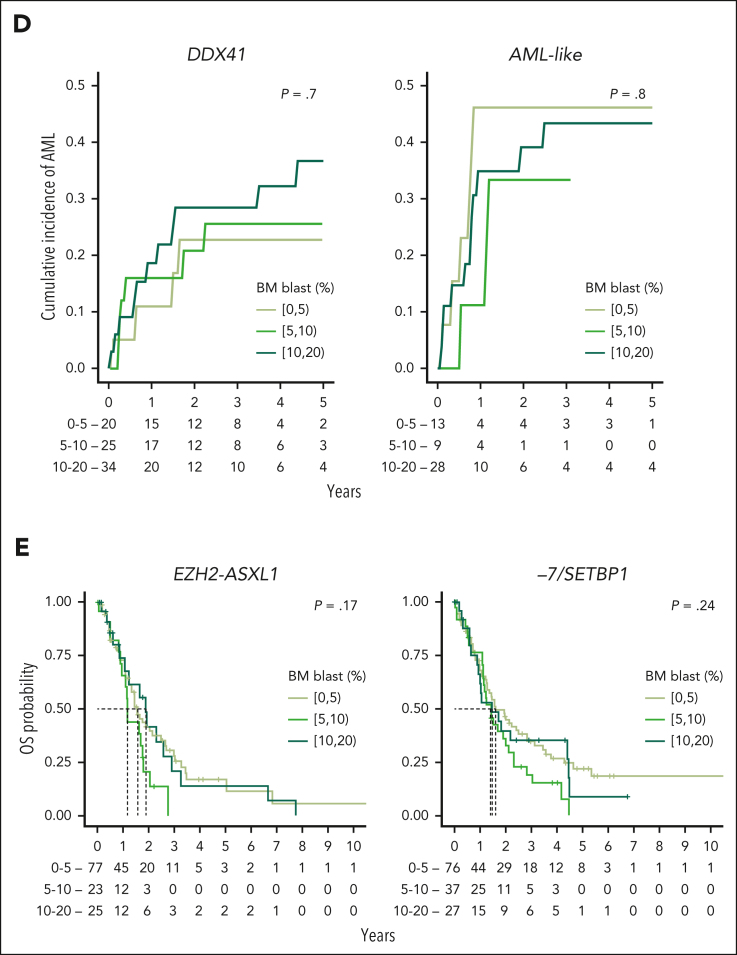
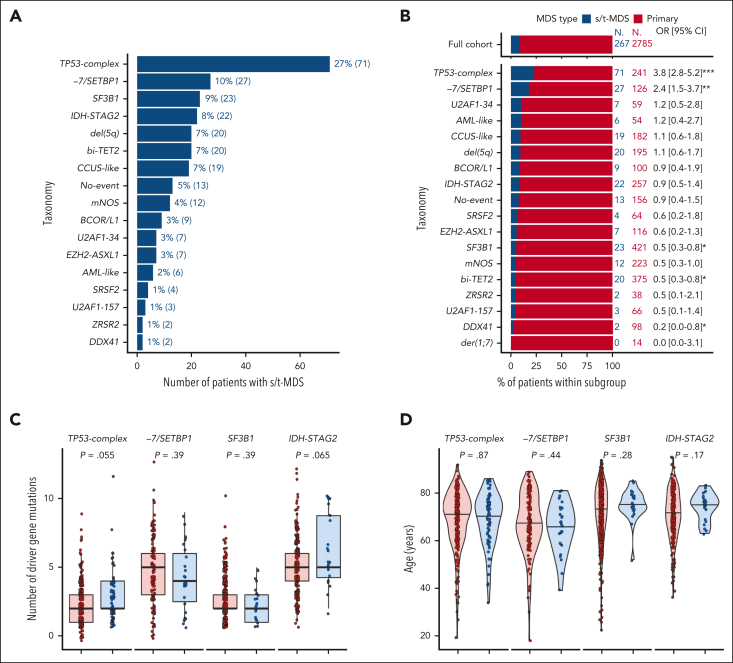
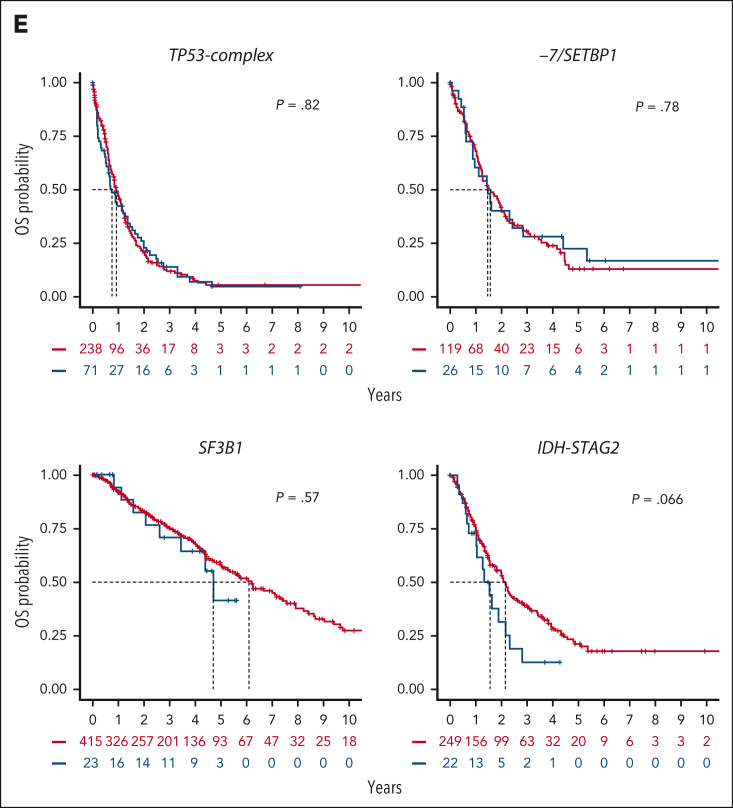
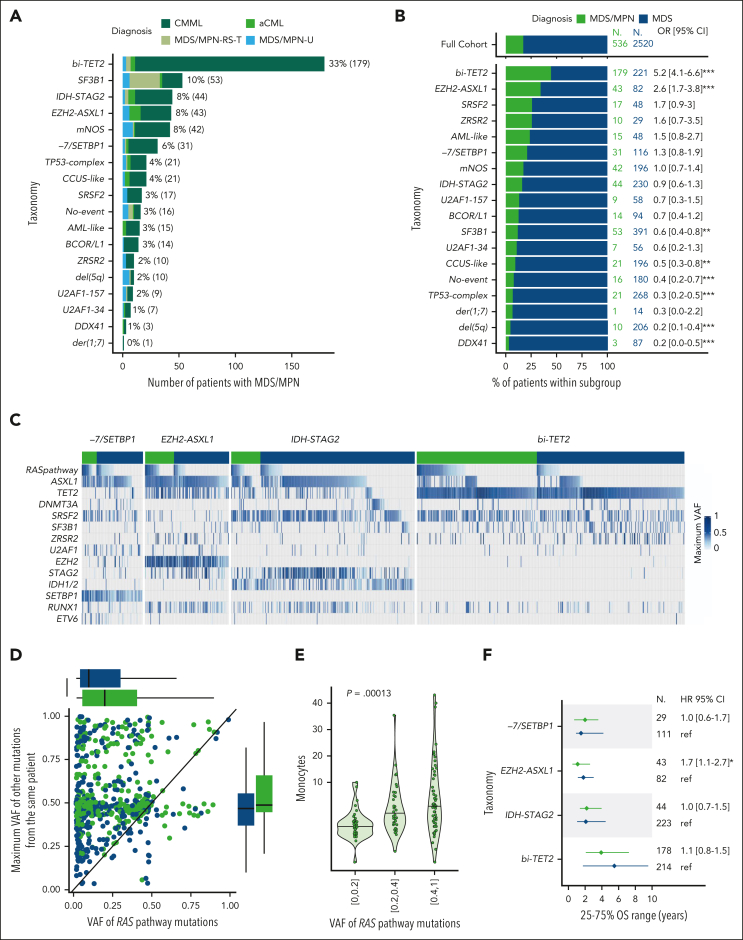
Myelodysplastic syndromes (MDS) are clonal hematologic disorders characterized by morphologic abnormalities of myeloid cells and peripheral cytopenias. Although genetic abnormalities underlie the pathogenesis of these disorders and their heterogeneity, current classifications of MDS rely predominantly on morphology. We performed genomic profiling of 3233 patients with MDS or related disorders to delineate molecular subtypes and define their clinical implications. Gene mutations, copy-number alterations, and copy-neutral loss of heterozygosity were derived from targeted sequencing of a 152-gene panel, with abnormalities identified in 91%, 43%, and 11% of patients, respectively. We characterized 16 molecular groups, encompassing 86% of patients, using information from 21 genes, 6 cytogenetic events, and loss of heterozygosity at the TP53 and TET2 loci. Two residual groups defined by negative findings (molecularly not otherwise specified, absence of recurrent drivers) comprised 14% of patients. The groups varied in size from 0.5% to 14% of patients and were associated with distinct clinical phenotypes and outcomes. The median bone marrow (BM) blast percentage across groups ranged from 1.5% to 10%, and the median overall survival ranged from 0.9 to 8.2 years. We validated 5 well-characterized entities, added further evidence to support 3 previously reported subsets, and described 8 novel groups. The prognostic influence of BM blasts depended on the genetic subtypes. Within genetic subgroups, therapy-related MDS and myelodysplastic/myeloproliferative neoplasms had comparable clinical and outcome profiles to primary MDS. In conclusion, genetically-derived subgroups of MDS are clinically relevant and might inform future classification schemas and translational therapeutic research.
© 2024 American Society of Hematology. Published by Elsevier Inc. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: A.A.v.d.L. reports research funding from Roche, Bristol Myers Squibb (BMS), and Celgene; and reports membership on the board of directors or advisory committees of BMS and Celgene. F.T. reports membership on the board of directors of Novartis and AbbVie. M.G.D.P. reports honoraria from, and membership on the board of directors or advisory committees of BMS. P.F. reports consultancy with, and honoraria from and research funding from Novartis, AbbVie, Janssen, Jazz Pharmaceuticals, and BMS; and reports honoraria from French MDS Group. M.R.S. reports membership on the board of directors or advisory committees of Savona, AbbVie Inc, BMS, Geron Corporation, Forma Therapeutics, CTI BioPharma Corp, Karyopharm Therapeutics Inc, Novartis, Sierra Oncology, Inc, Taiho, Takeda Pharmaceutical, and TG Therapeutics Inc; reports consultancy with Forma Therapeutics Inc, Karyopharm Therapeutics Inc, Ryvu Therapeutics; is a current equity holder in publicly traded companies, Karyopharm Therapeutics Inc and Ryvu Therapeutics; received research funding from Takeda Pharmaceutical, TG Therapeutics Inc, ALX Oncology, Astex Pharmaceuticals, Incyte Corporation; and reports patents with, and royalties from, Boehringer Ingelheim. P.V. reports honoraria from BMS, Novartis, Pfizer, Incyte, Blueprint, and Stemline. I.K. reports consultancy with Novartis, BMS, Genesis, and AbbVie; received research funding from Novartis; and received honoraria from Genesis and AbbVie. V.S. reports membership on the board of directors or advisory committees of Santini, BMS, AbbVie, Geron, Gilead, CTI BioPharma, Otsuka, Servier, Janssen, and Syros. U.P. received research funding from Janssen Biotech, Geron, Fibrogen, Silence Therapeutics, Takeda, Curis, Merck, Servier, Syros, Novartis, Celgene, BMS, Amgen, Roche, Jazz Pharmaceuticals, and BeiGene; serves in a consulting role for Janssen Biotech, Geron, Silence Therapeutics, Takeda, Curis, Servier, Jazz Pharmaceuticals, Syros, AbbVie, Novartis, BMS, and Amgen; received honoraria from Silence Therapeutics, Celgene, Takeda, Servier, Jazz Pharmaceuticals, Syros, Novartis, and BMS; reports membership on the board of directors or advisory committees of MDS Foundation and BMS; and received other support (travel support and medical writing) from BMS. M.H. reports consultancy with Pfizer, PinotBio, AbbVie, Servier, BMS, Glycostem, Jazz Pharmaceuticals, Amgen, and Delbert Lab; received honoraria from Pfizer, Novartis, Sobi, Certara, Janssen, and Jazz Pharmaceuticals; and received research funding from PinotBio, BerGenBio, Astellas, Agios, AbbVie, Loxo Oncology, BMS, Glycostem, Karyopharm, and Jazz Pharmaceuticals. M.T.V. serves on the advisory board of Jazz Pharmaceuticals, Celgene/BMS, and Syros; received research funding from Novartis and Celgene/BMS; and is a member of the speakers bureau of AstraZeneca, Novartis, AbbVie, Jazz Pharmaceuticals, Astellas, and Celgene/BMS. B.L.E. is a current equity holder in private company, Skyhawk Therapeutics, Exo Therapeutics, TenSixteen Bio, and Neomorph Inc; reports membership on the board of directors or advisory committees of Skyhawk Therapeutics, Exo Therapeutics, TenSixteen Bio, and Neomorph Inc; serves in a consulting role for AbbVie; and received research funding from Calico and Novartis. P.L.G. reports consultancy with, and research funding from BMS, Novartis, and Gilead. N.G. received research funding from Takeda; and honoraria from Novartis and BMS. E.P. is cofounder of, and holds a fiduciary role in a private company, Isabl Inc; and holds stock options in a private company, TenSixteen Bio. The remaining authors declare no competing financial interests.
Figures

Comment in
-
Seeing MDS through the lens of genomics.Blood. 2024 Oct 10;144(15):1552-1554. doi: 10.1182/blood.2024025676. Blood. 2024. PMID: 39388160 No abstract available.
References
-
- Cazzola M. Myelodysplastic syndromes. N Engl J Med. 2020;383(14):1358–1374. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
