

Downstream-of-gene (DoG) transcripts contribute to an imbalance in the cancer cell transcriptome
- PMID: 38959318
- PMCID: PMC11221514
- DOI: 10.1126/sciadv.adh9613
Downstream-of-gene (DoG) transcripts contribute to an imbalance in the cancer cell transcriptome
Abstract
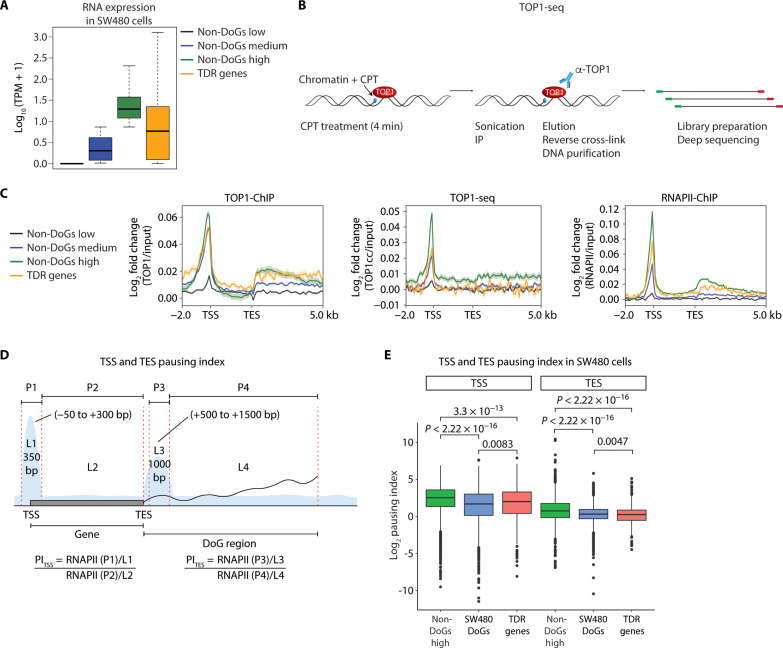
Downstream-of-gene (DoG) transcripts are an emerging class of noncoding RNAs. However, it remains largely unknown how DoG RNA production is regulated and whether alterations in DoG RNA signatures exist in major cancers. Here, through transcriptomic analyses of matched tumors and nonneoplastic tissues and cancer cell lines, we reveal a comprehensive catalog of DoG RNA signatures. Through separate lines of evidence, we support the biological importance of DoG RNAs in carcinogenesis. First, we show tissue-specific and stage-specific differential expression of DoG RNAs in tumors versus paired normal tissues with their respective host genes involved in tumor-promoting versus tumor-suppressor pathways. Second, we identify that differential DoG RNA expression is associated with poor patient survival. Third, we identify that DoG RNA induction is a consequence of treating colon cancer cells with the topoisomerase I (TOP1) poison camptothecin and following TOP1 depletion. Our results underlie the significance of DoG RNAs and TOP1-dependent regulation of DoG RNAs in diversifying and modulating the cancer transcriptome.
Figures






References
-
- Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
