

A unified metric of human immune health
- PMID: 38961223
- PMCID: PMC12183718
- DOI: 10.1038/s41591-024-03092-6
A unified metric of human immune health
Abstract
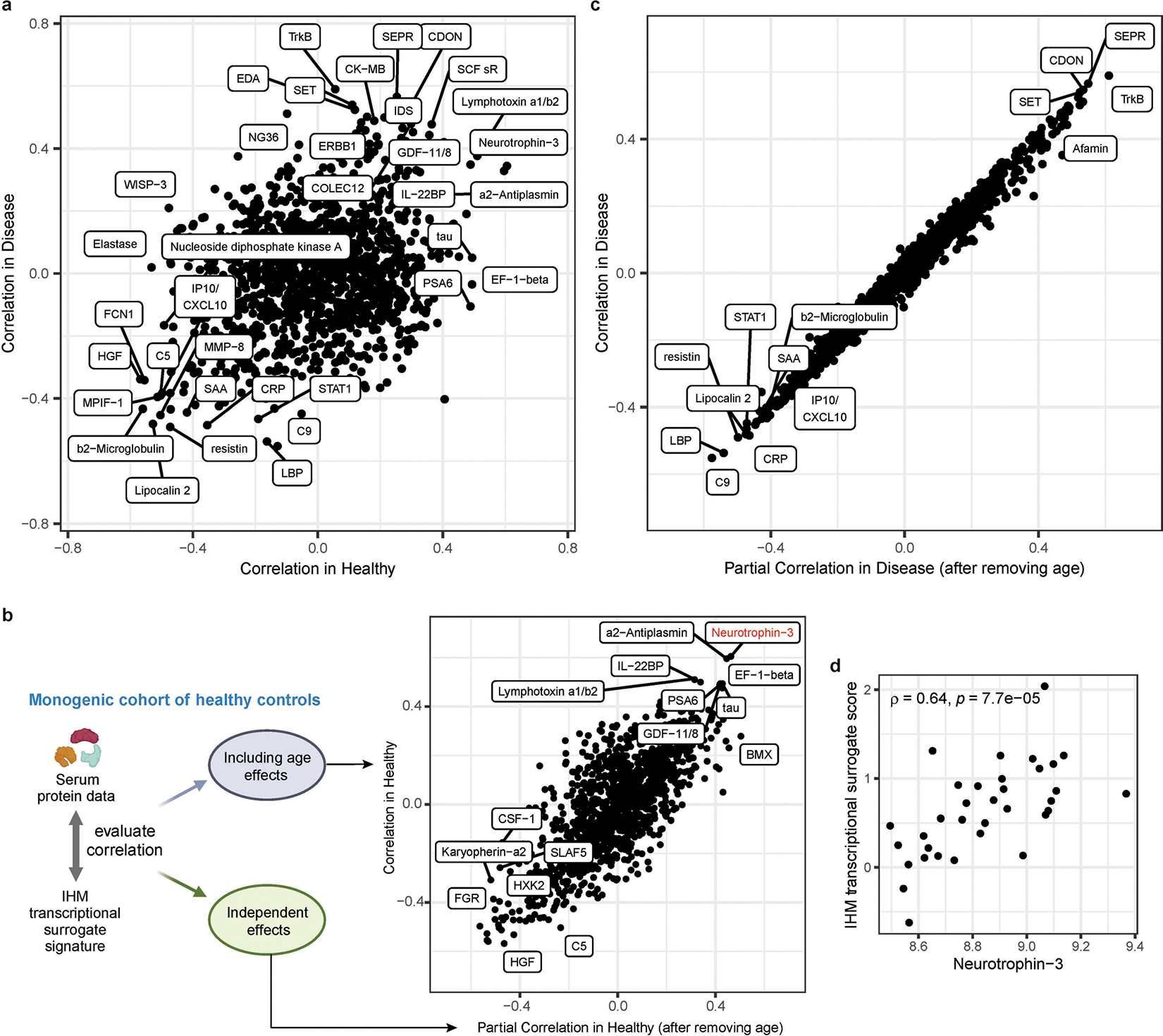
Immunological health has been challenging to characterize but could be defined as the absence of immune pathology. While shared features of some immune diseases and the concept of immunologic resilience based on age-independent adaptation to antigenic stimulation have been developed, general metrics of immune health and its utility for assessing clinically healthy individuals remain ill defined. Here we integrated transcriptomics, serum protein, peripheral immune cell frequency and clinical data from 228 patients with 22 monogenic conditions impacting key immunological pathways together with 42 age- and sex-matched healthy controls. Despite the high penetrance of monogenic lesions, differences between individuals in diverse immune parameters tended to dominate over those attributable to disease conditions or medication use. Unsupervised or supervised machine learning independently identified a score that distinguished healthy participants from patients with monogenic diseases, thus suggesting a quantitative immune health metric (IHM). In ten independent datasets, the IHM discriminated healthy from polygenic autoimmune and inflammatory disease states, marked aging in clinically healthy individuals, tracked disease activities and treatment responses in both immunological and nonimmunological diseases, and predicted age-dependent antibody responses to immunizations with different vaccines. This discriminatory power goes beyond that of the classical inflammatory biomarkers C-reactive protein and interleukin-6. Thus, deviations from health in diverse conditions, including aging, have shared systemic immune consequences, and we provide a web platform for calculating the IHM for other datasets, which could empower precision medicine.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
J.S.T. serves on the Scientific Advisory Boards of CytoReason, Inc., the Human Immunome Project and ImmunoScape. B.A.S. is a former SomaLogic, Inc. (Boulder, CO, USA) employee and a company shareholder. S.H.K. receives consulting fees from Peraton. All other authors declare no competing interests.
Figures










Update of
-
Multiomics integration of 22 immune-mediated monogenic diseases reveals an emergent axis of human immune health.Res Sq [Preprint]. 2023 Mar 20:rs.3.rs-2070975. doi: 10.21203/rs.3.rs-2070975/v1. Res Sq. 2023. Update in: Nat Med. 2024 Sep;30(9):2461-2472. doi: 10.1038/s41591-024-03092-6. PMID: 36993430 Free PMC article. Updated. Preprint.
References
-
- Medzhitov R The spectrum of inflammatory responses. Science 374, 1070–1075 (2021). - PubMed
-
- Leonard WJ, Lin J-X & O’Shea JJ The γc family of cytokines: basic biology to therapeutic ramifications. Immunity 50, 832–850 (2019). - PubMed
-
- Manthiram K, Zhou Q, Aksentijevich I & Kastner DL The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol. 18, 832–842 (2017). - PubMed
MeSH terms
Substances
Grants and funding
- ZIA AI001152/ImNIH/Intramural NIH HHS/United States
- 75N91019D00024/CA/NCI NIH HHS/United States
- U19AI089992/U.S. Department of Health & Human Services | NIH | Office of Extramural Research, National Institutes of Health (OER)
- Z-01-A-00646/Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Division of Intramural Research of the NIAID)
- U19 AI145825/AI/NIAID NIH HHS/United States
- R01 AI170116/AI/NIAID NIH HHS/United States
- 1ZIAAI001152/Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Division of Intramural Research of the NIAID)
- 1 ZIA AI001202-07/Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Division of Intramural Research of the NIAID)
- 5R01AI170116/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)
- 1ZIAHG200371/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- Z01 AI000825-24 LIR/Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Division of Intramural Research of the NIAID)
- Z99 AG999999/ImNIH/Intramural NIH HHS/United States
- ZIA-HL006089-12/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- 1ZIAHG200374/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- Z-01-A-00647/Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Division of Intramural Research of the NIAID)
- 1ZIAHG200373/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- U19 AI089992/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
