

A µ-opioid receptor modulator that works cooperatively with naloxone
- PMID: 38961287
- PMCID: PMC11998091
- DOI: 10.1038/s41586-024-07587-7
A µ-opioid receptor modulator that works cooperatively with naloxone
Abstract
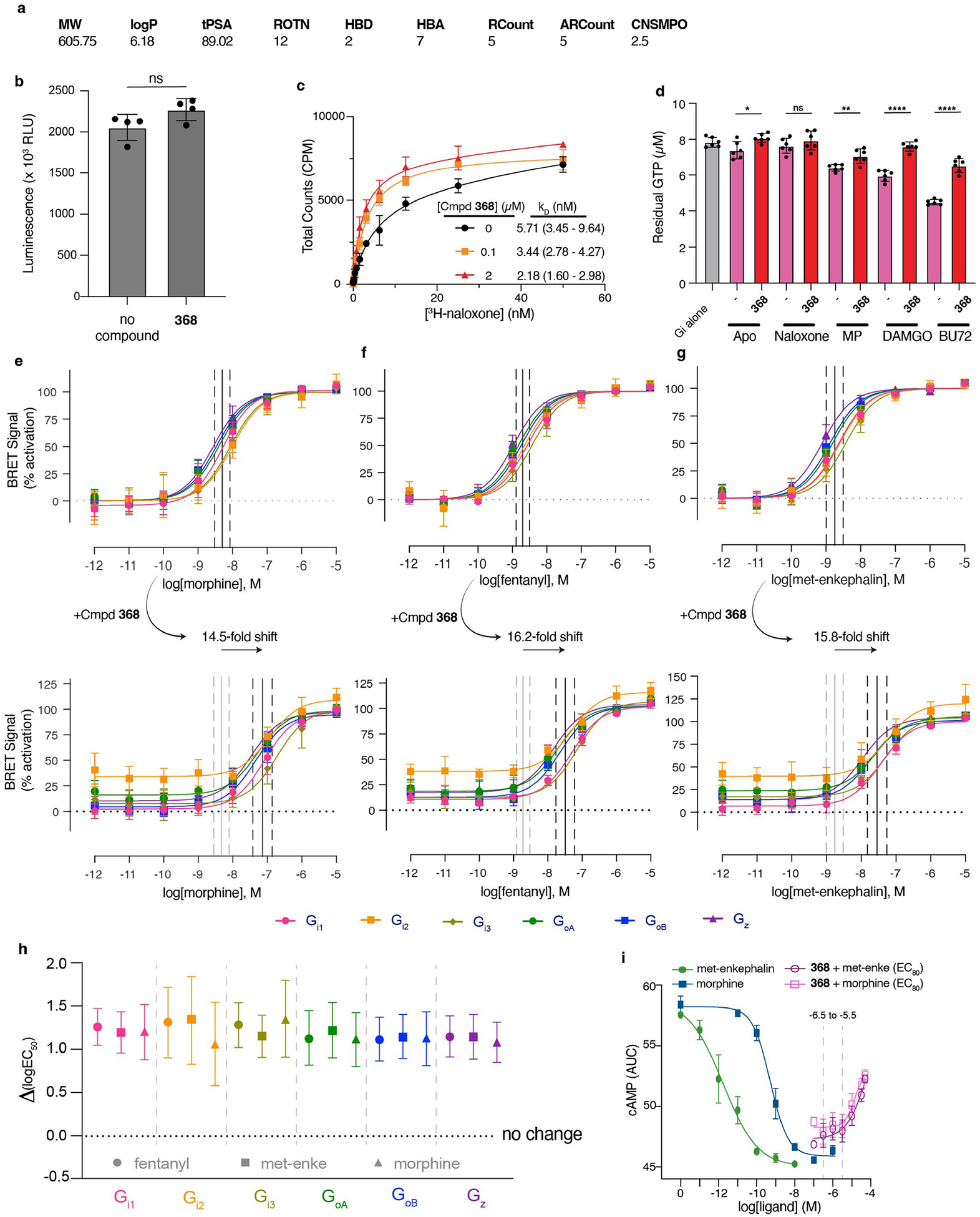
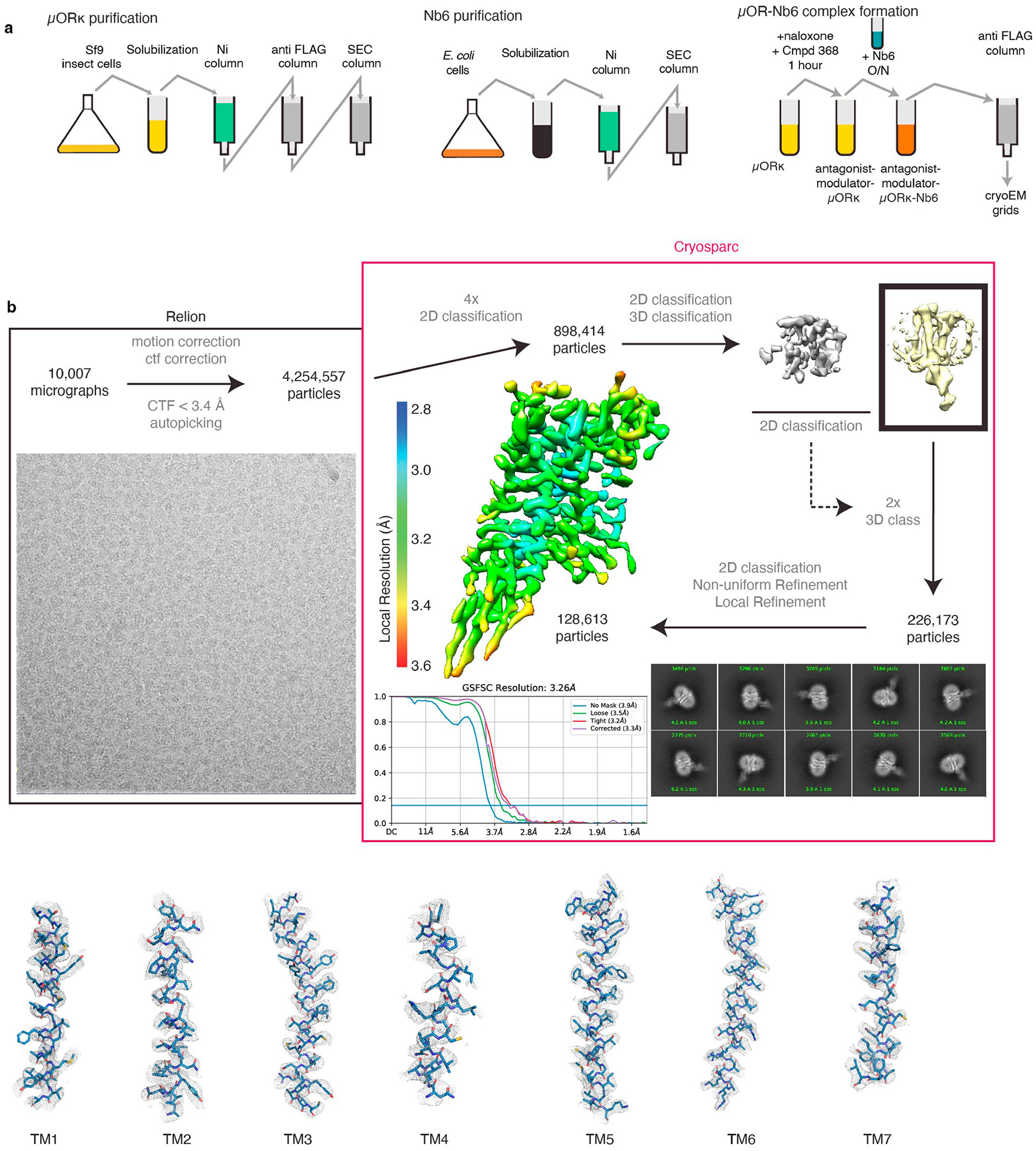
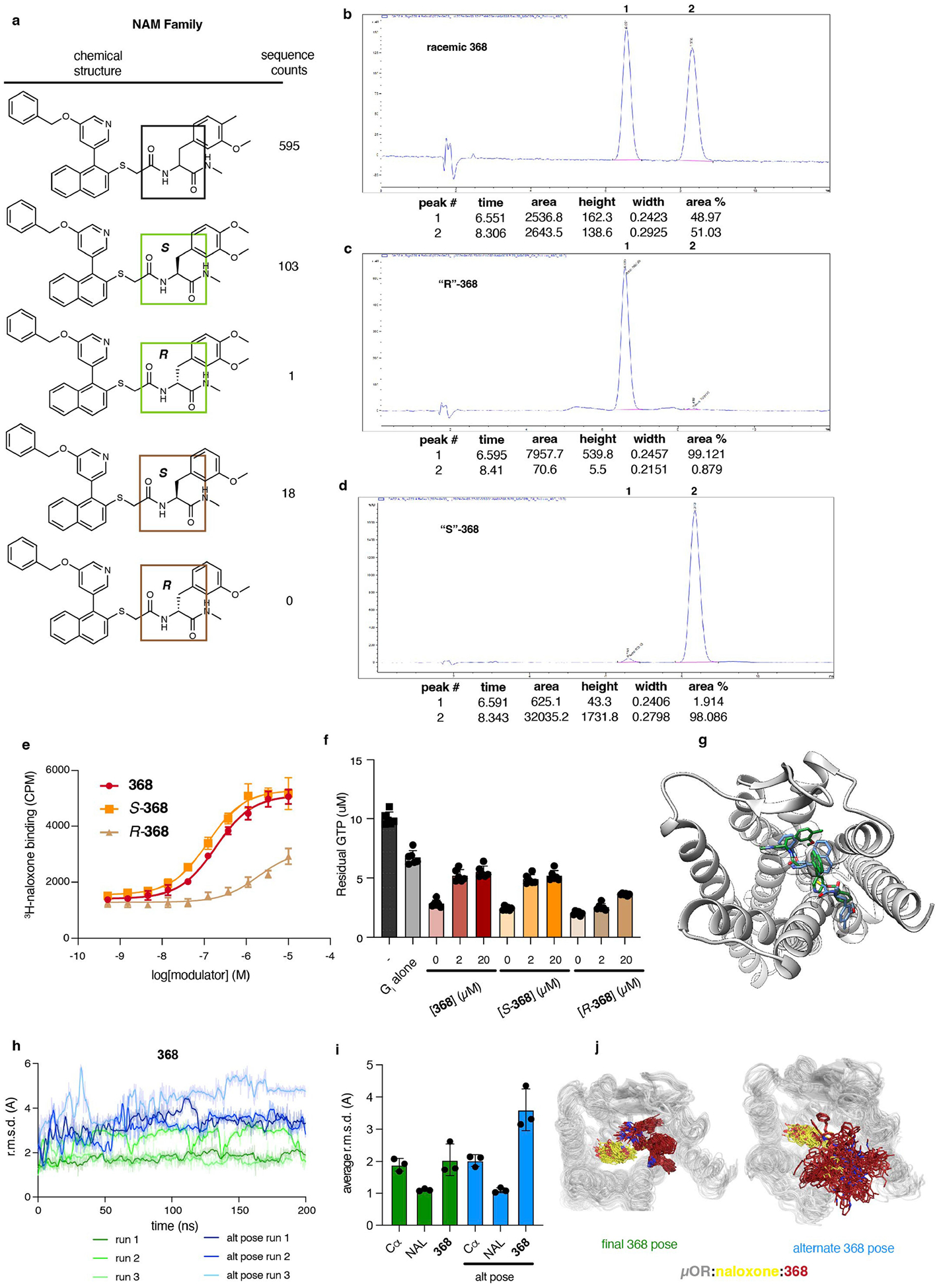
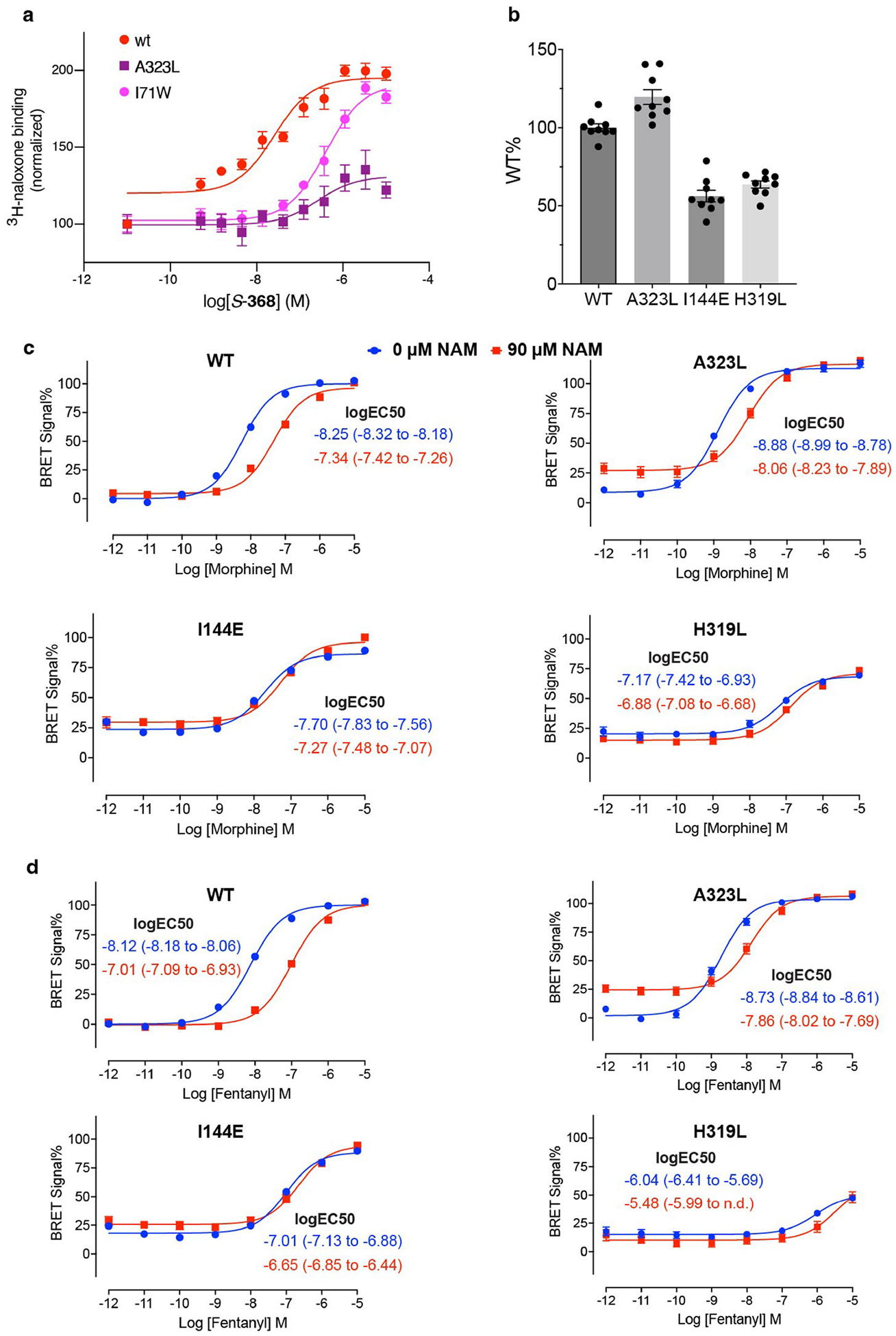
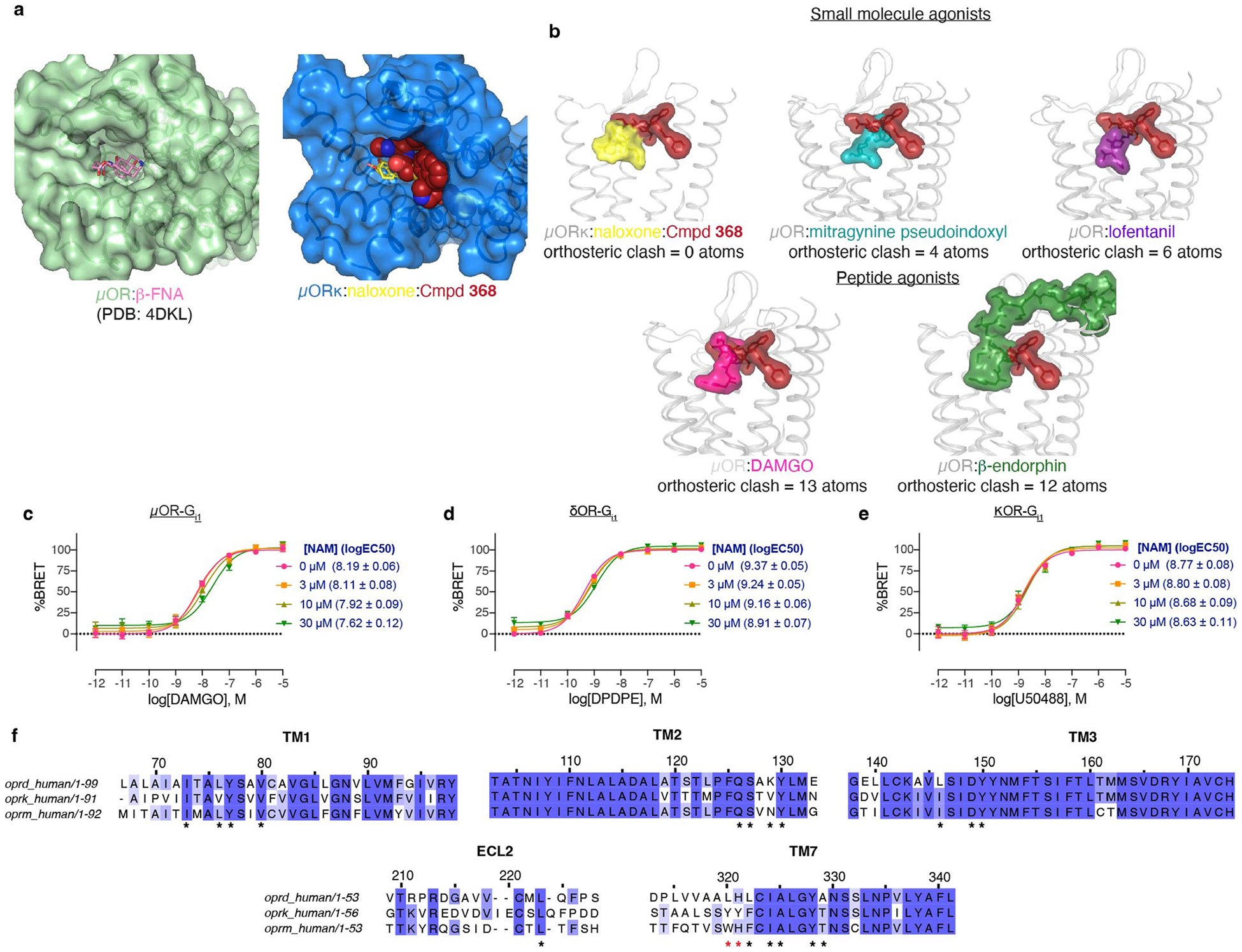
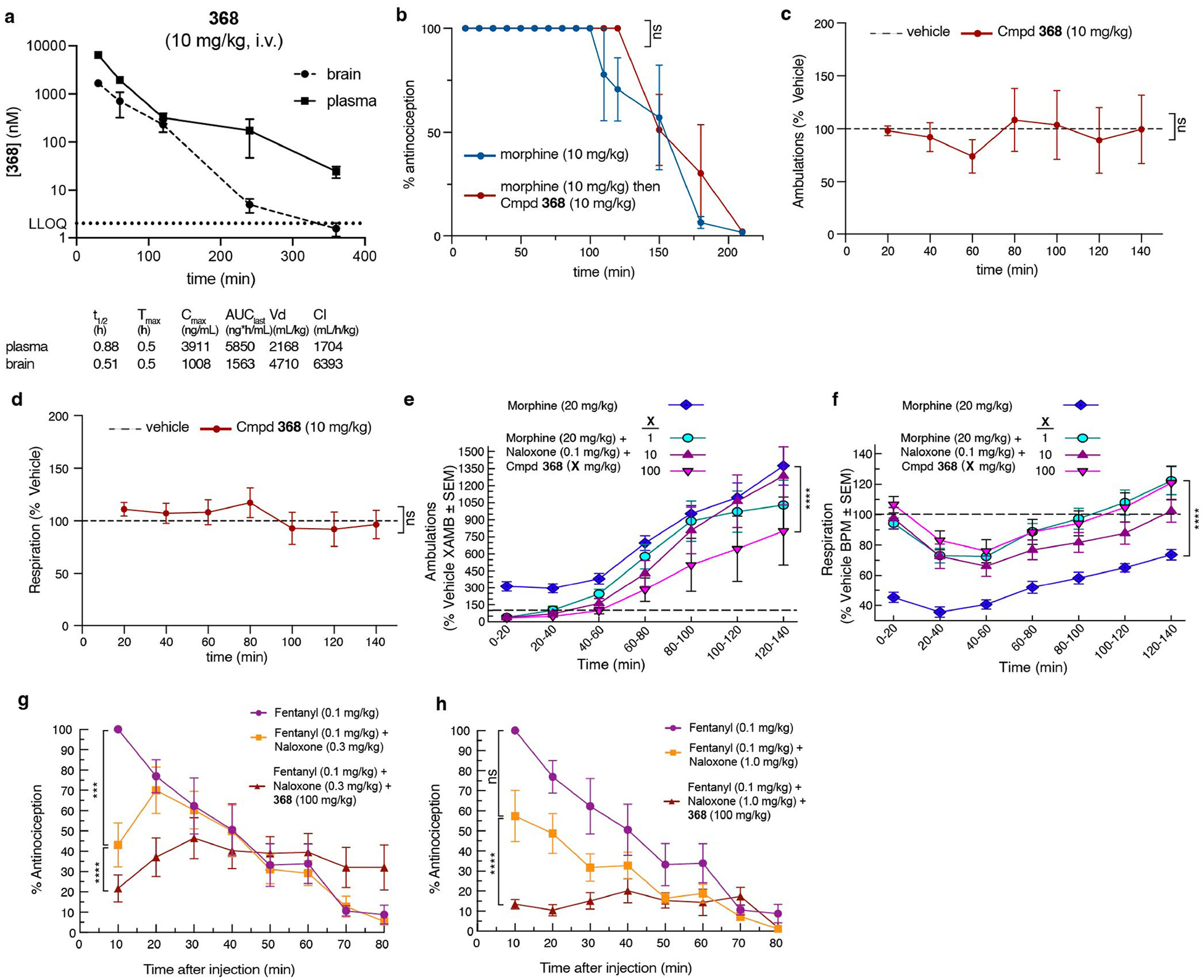
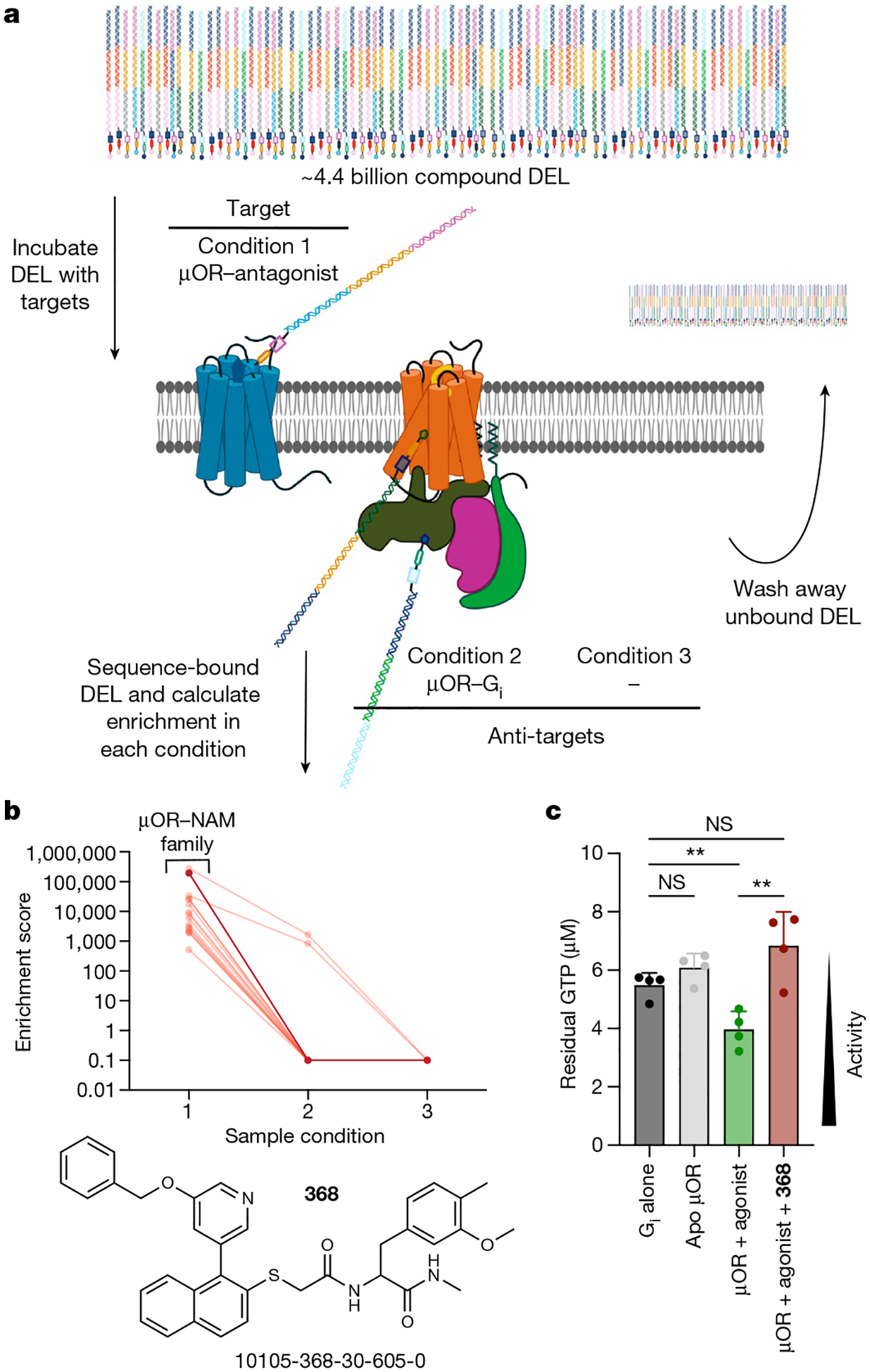
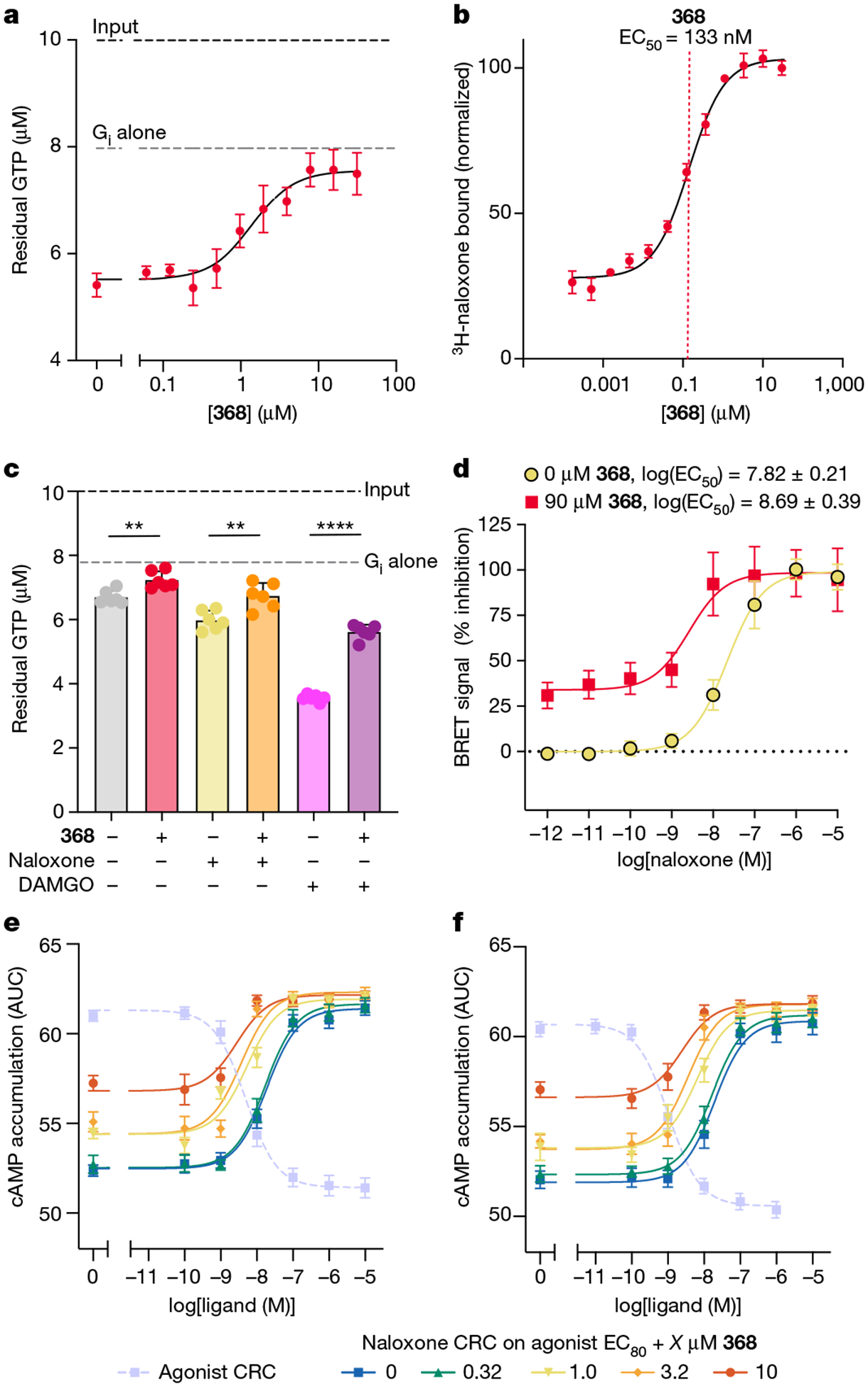
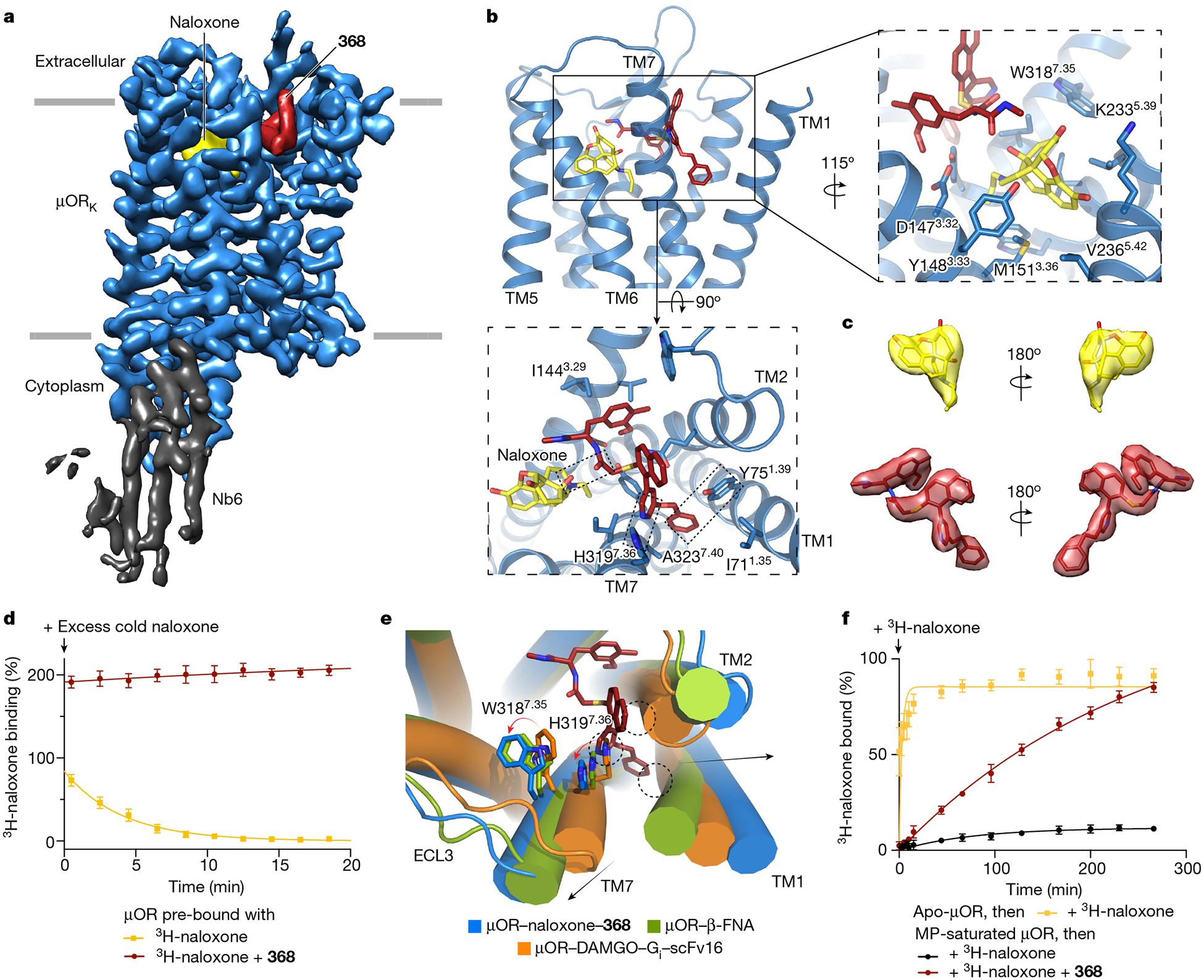
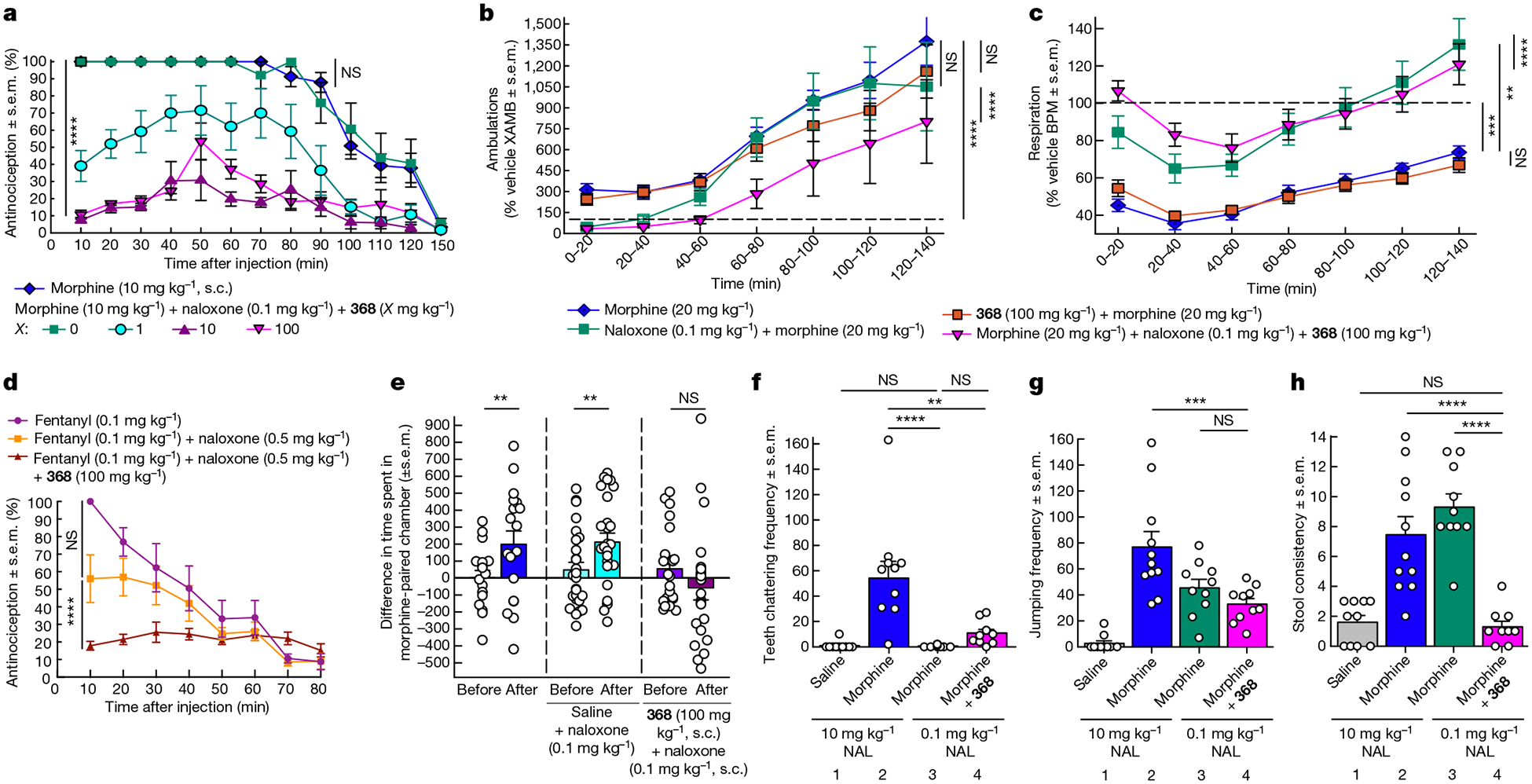
The µ-opioid receptor (µOR) is a well-established target for analgesia1, yet conventional opioid receptor agonists cause serious adverse effects, notably addiction and respiratory depression. These factors have contributed to the current opioid overdose epidemic driven by fentanyl2, a highly potent synthetic opioid. µOR negative allosteric modulators (NAMs) may serve as useful tools in preventing opioid overdose deaths, but promising chemical scaffolds remain elusive. Here we screened a large DNA-encoded chemical library against inactive µOR, counter-screening with active, G-protein and agonist-bound receptor to 'steer' hits towards conformationally selective modulators. We discovered a NAM compound with high and selective enrichment to inactive µOR that enhances the affinity of the key opioid overdose reversal molecule, naloxone. The NAM works cooperatively with naloxone to potently block opioid agonist signalling. Using cryogenic electron microscopy, we demonstrate that the NAM accomplishes this effect by binding a site on the extracellular vestibule in direct contact with naloxone while stabilizing a distinct inactive conformation of the extracellular portions of the second and seventh transmembrane helices. The NAM alters orthosteric ligand kinetics in therapeutically desirable ways and works cooperatively with low doses of naloxone to effectively inhibit various morphine-induced and fentanyl-induced behavioural effects in vivo while minimizing withdrawal behaviours. Our results provide detailed structural insights into the mechanism of negative allosteric modulation of the µOR and demonstrate how this can be exploited in vivo.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures
Comment in
-
Opioid antidote gets a boost.Nat Rev Drug Discov. 2024 Sep;23(9):658. doi: 10.1038/d41573-024-00125-0. Nat Rev Drug Discov. 2024. PMID: 39054396 No abstract available.
References
-
- Jamison RN & Mao J Opioid analgesics. Mayo Clin. Proc 90, 957–968 (2015). - PubMed
-
- Centers for Disease Control and Prevention. Vital signs: overdoses of prescription opioid pain relievers—United States, 1999–2008. MMWR Morb. Mortal. Wkly Rep 60, 1487–1492 (2011). - PubMed
-
- National Academies of Sciences, Engineering, and Medicine. Pain Management and the Opioid Epidemic: Balancing Societal and Individual Benefits and Risks of Prescription Opioid Use (National Academies Press, 2017). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
