

Geographical migration and fitness dynamics of Streptococcus pneumoniae
- PMID: 38961295
- PMCID: PMC11236706
- DOI: 10.1038/s41586-024-07626-3
Geographical migration and fitness dynamics of Streptococcus pneumoniae
Abstract
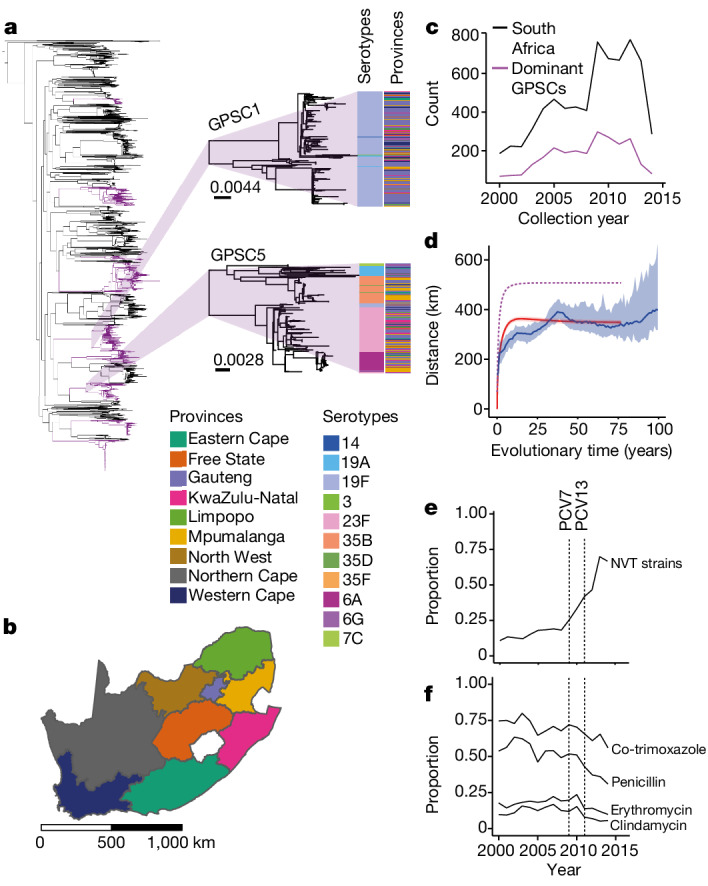
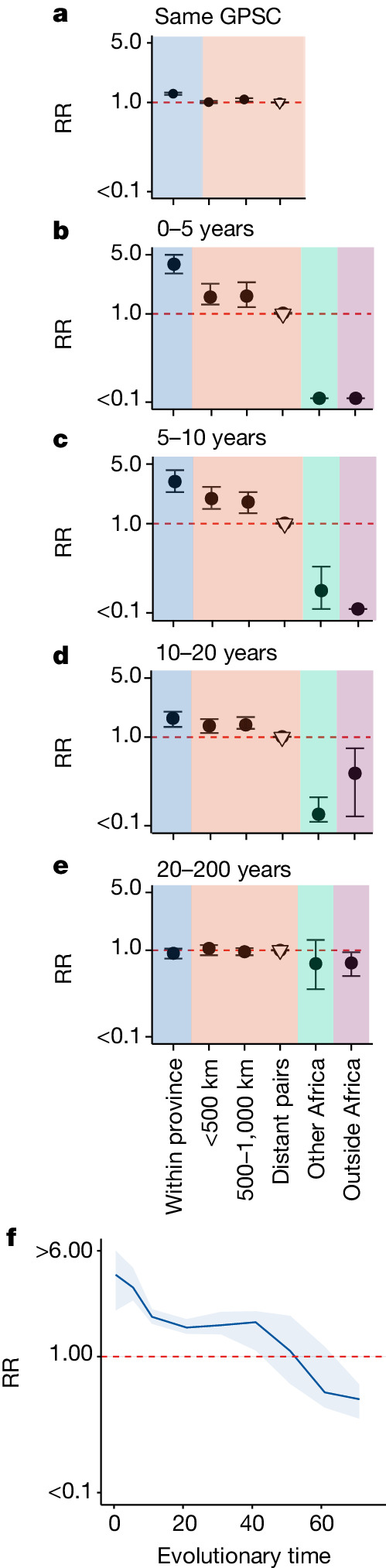
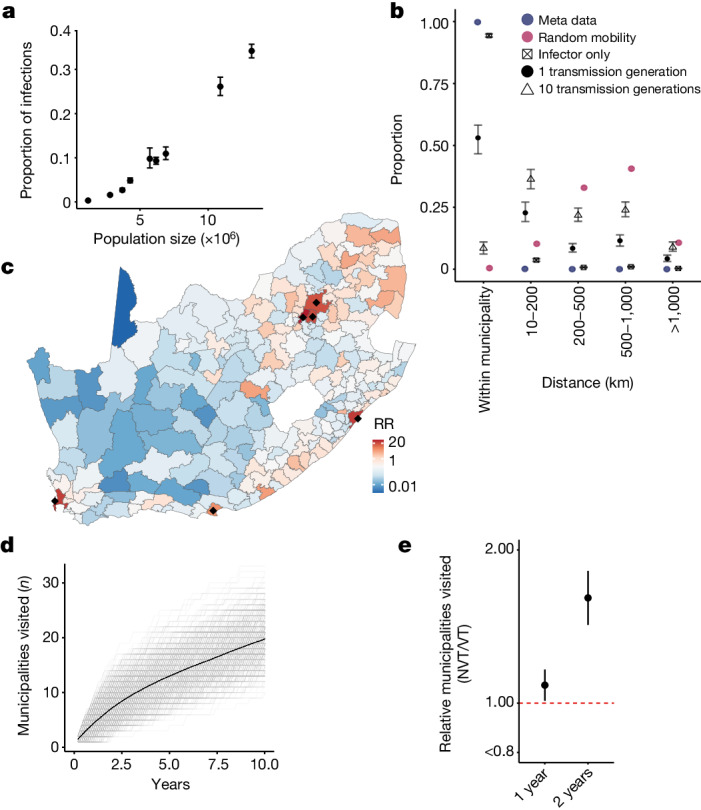
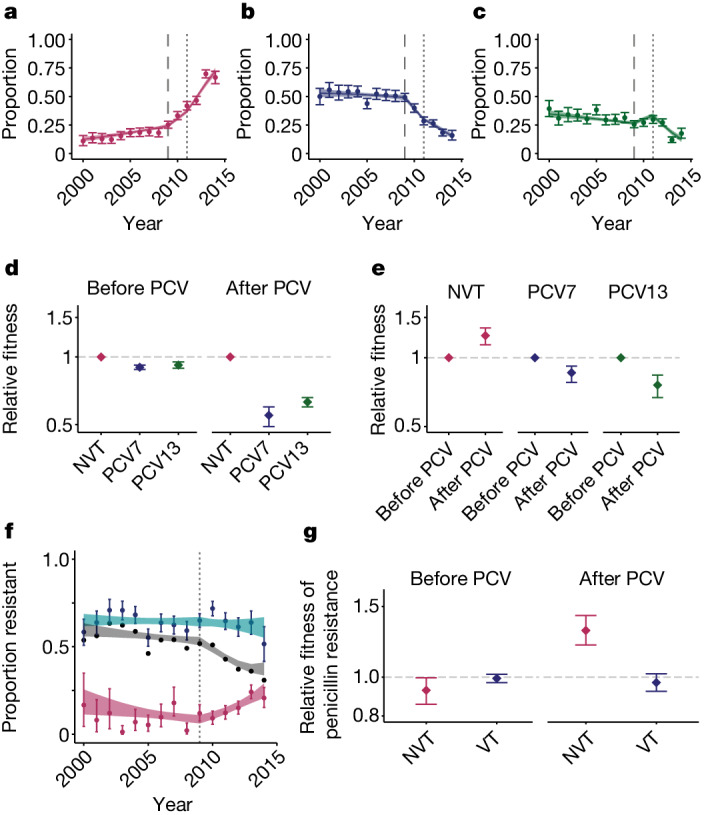
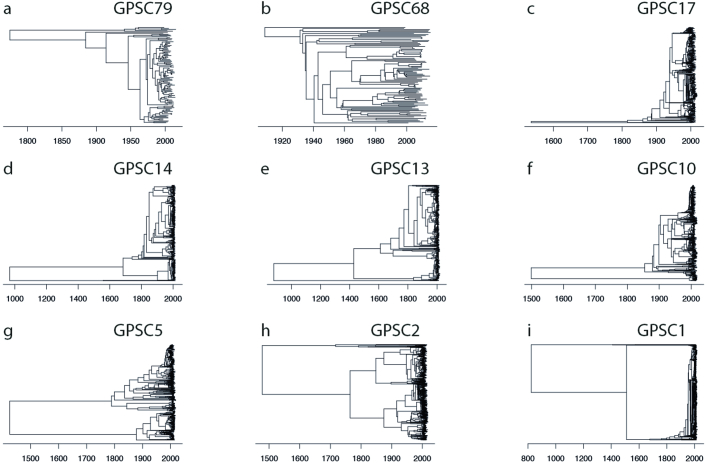
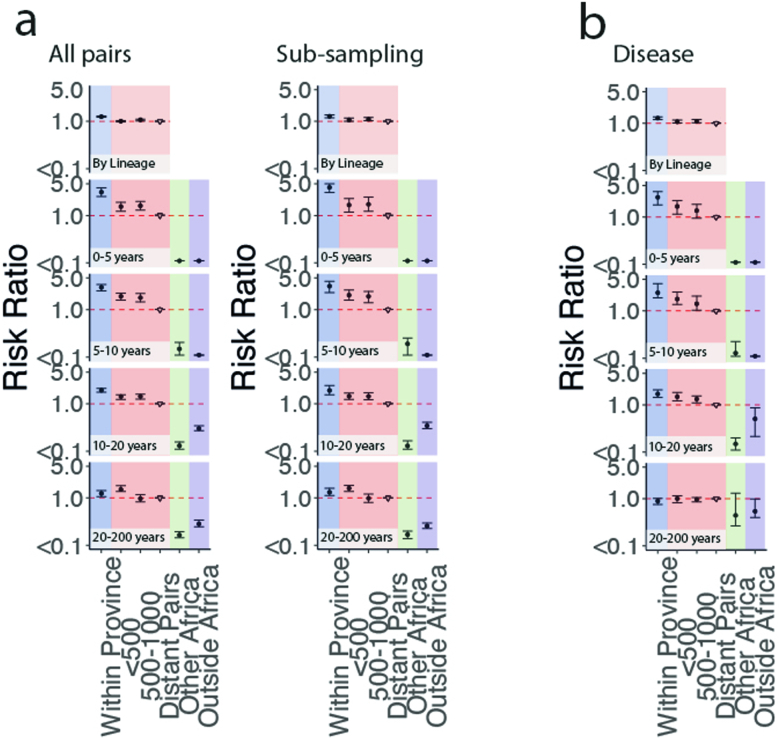
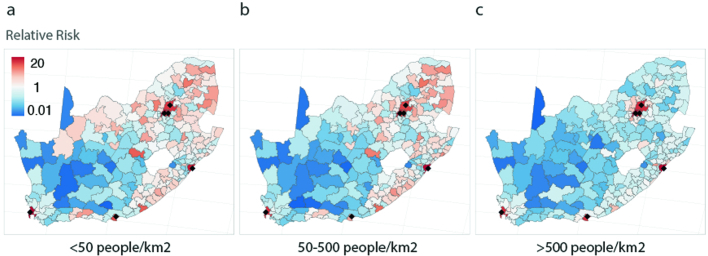
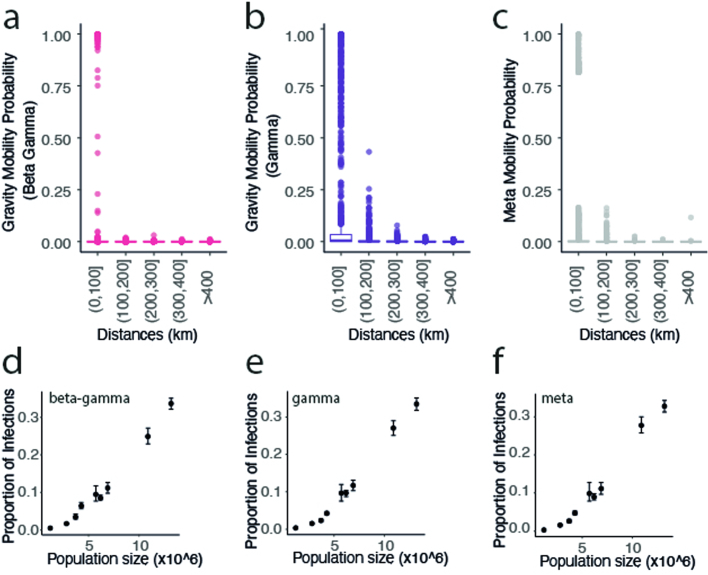
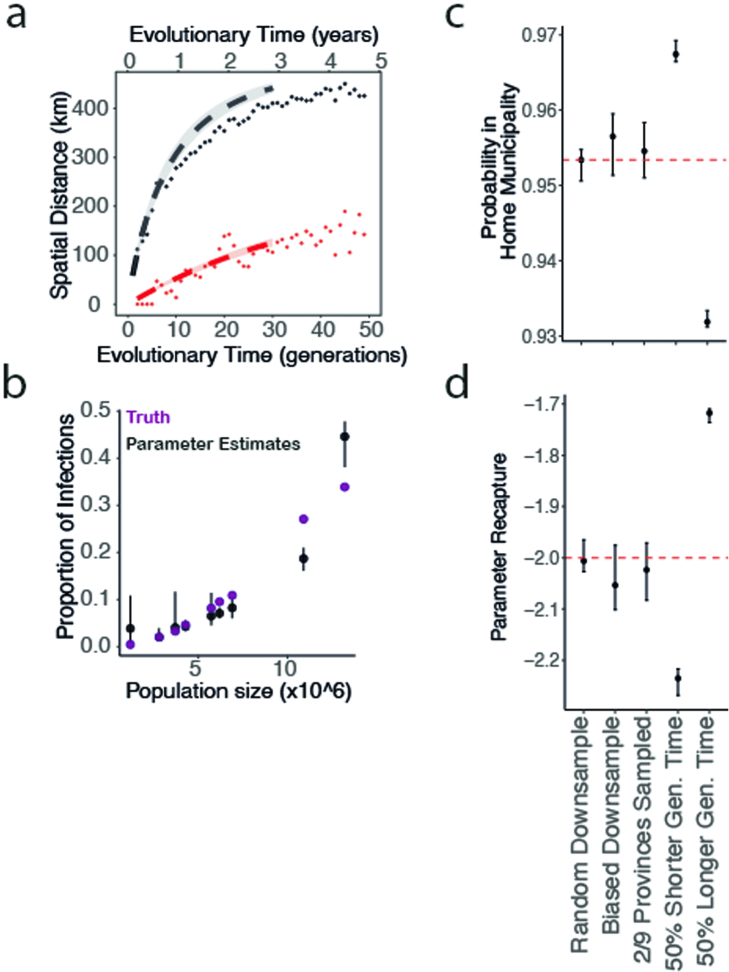
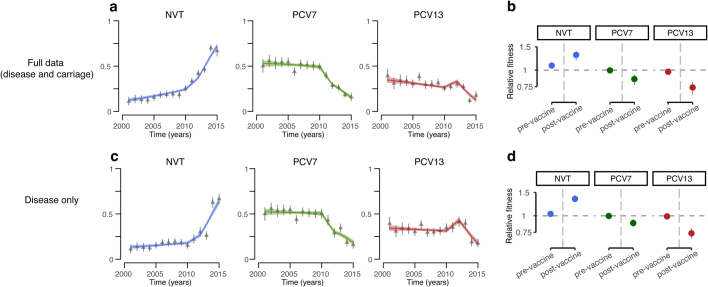
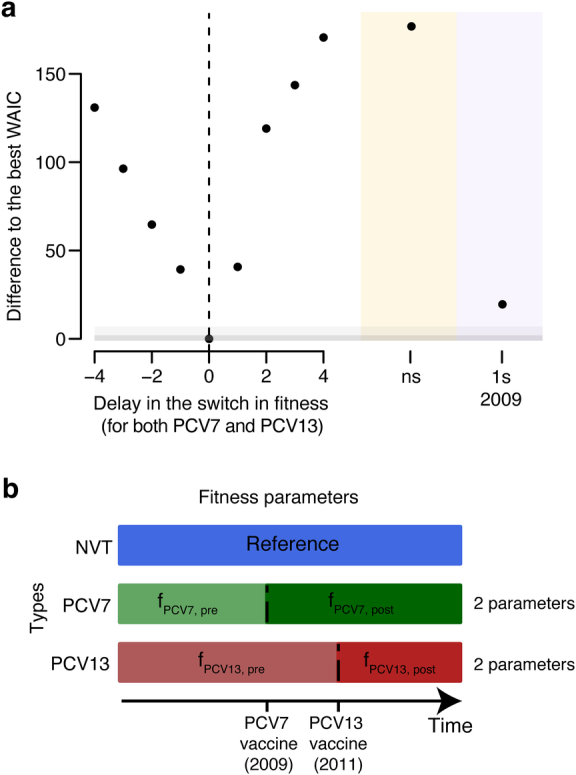
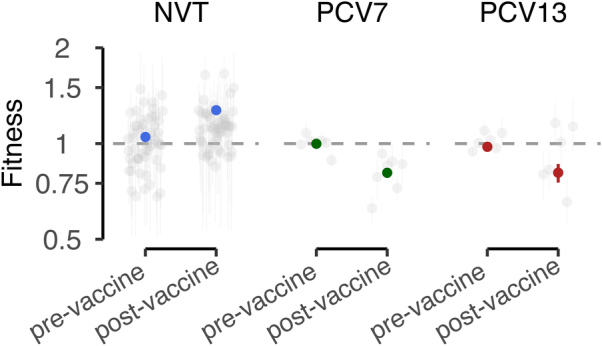
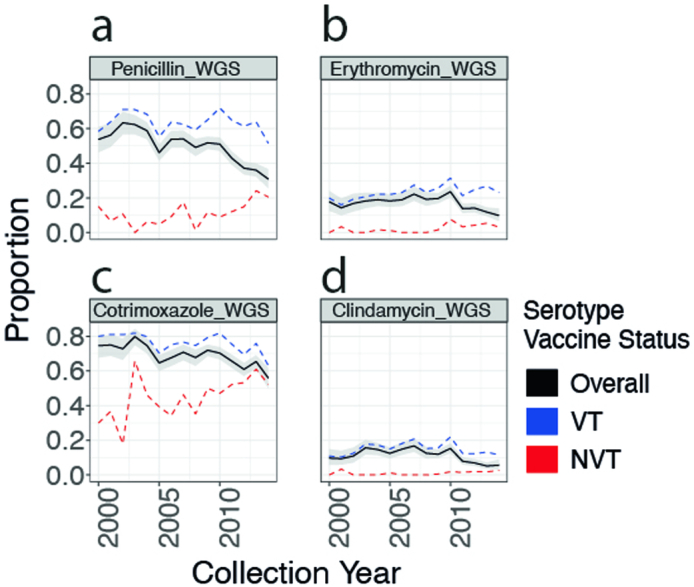
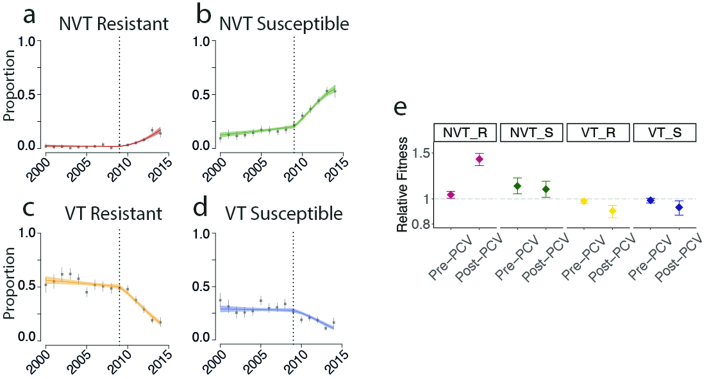
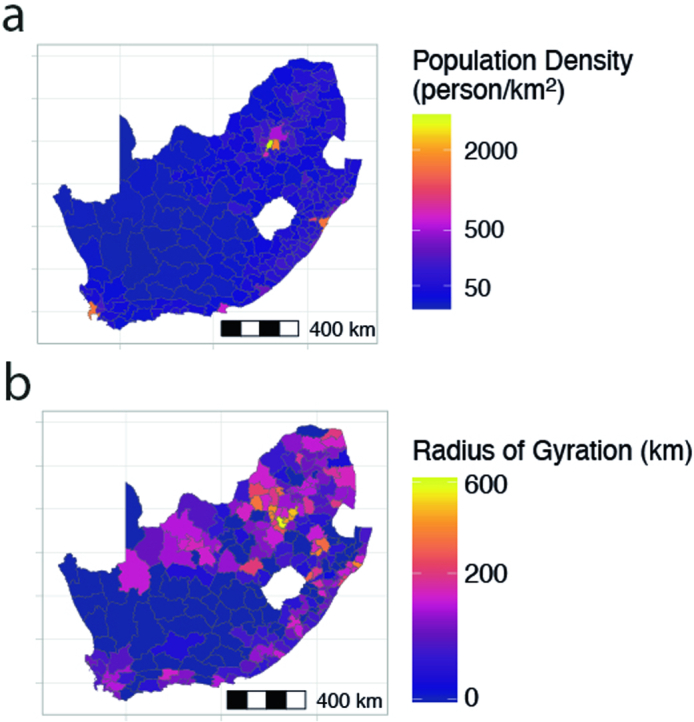
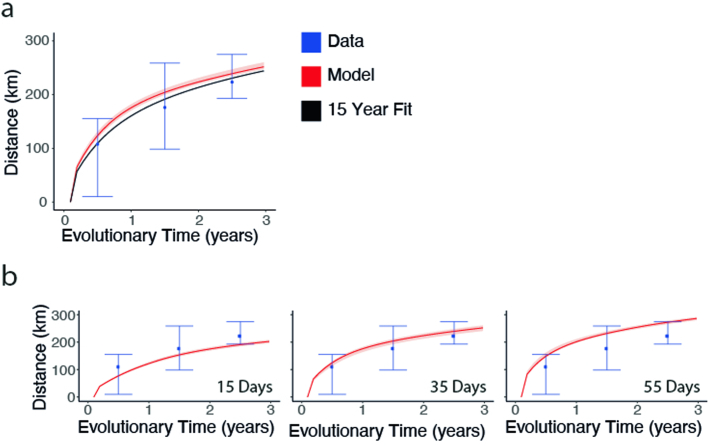
Streptococcus pneumoniae is a leading cause of pneumonia and meningitis worldwide. Many different serotypes co-circulate endemically in any one location1,2. The extent and mechanisms of spread and vaccine-driven changes in fitness and antimicrobial resistance remain largely unquantified. Here using geolocated genome sequences from South Africa (n = 6,910, collected from 2000 to 2014), we developed models to reconstruct spread, pairing detailed human mobility data and genomic data. Separately, we estimated the population-level changes in fitness of strains that are included (vaccine type (VT)) and not included (non-vaccine type (NVT)) in pneumococcal conjugate vaccines, first implemented in South Africa in 2009. Differences in strain fitness between those that are and are not resistant to penicillin were also evaluated. We found that pneumococci only become homogenously mixed across South Africa after 50 years of transmission, with the slow spread driven by the focal nature of human mobility. Furthermore, in the years following vaccine implementation, the relative fitness of NVT compared with VT strains increased (relative risk of 1.68; 95% confidence interval of 1.59-1.77), with an increasing proportion of these NVT strains becoming resistant to penicillin. Our findings point to highly entrenched, slow transmission and indicate that initial vaccine-linked decreases in antimicrobial resistance may be transient.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- World Health Organization. The top 10 causes of death. WHOhttps://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (2020).
-
- Troeger C, et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 2017;17:1133–1161. doi: 10.1016/S1473-3099(17)30396-1. - DOI - PMC - PubMed
-
- Bender, R. G. et al. Global, regional, and national incidence and mortality burden of non-COVID-19 lower respiratory infections and aetiologies, 1990–2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Infect. Dis.10.1016/S1473-3099(24)00176-2 (2024). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
