

Spike deep mutational scanning helps predict success of SARS-CoV-2 clades
- PMID: 38961298
- PMCID: PMC11254757
- DOI: 10.1038/s41586-024-07636-1
Spike deep mutational scanning helps predict success of SARS-CoV-2 clades
Abstract
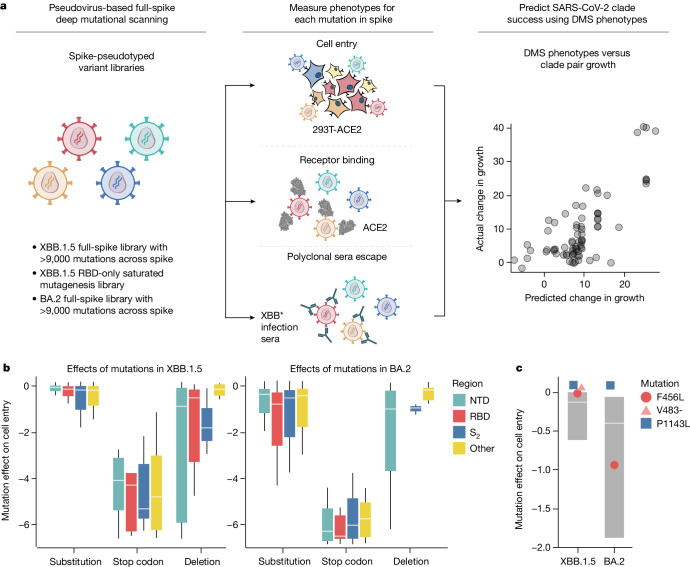
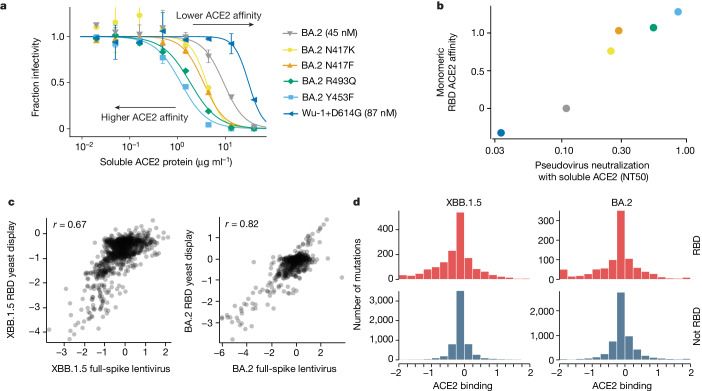
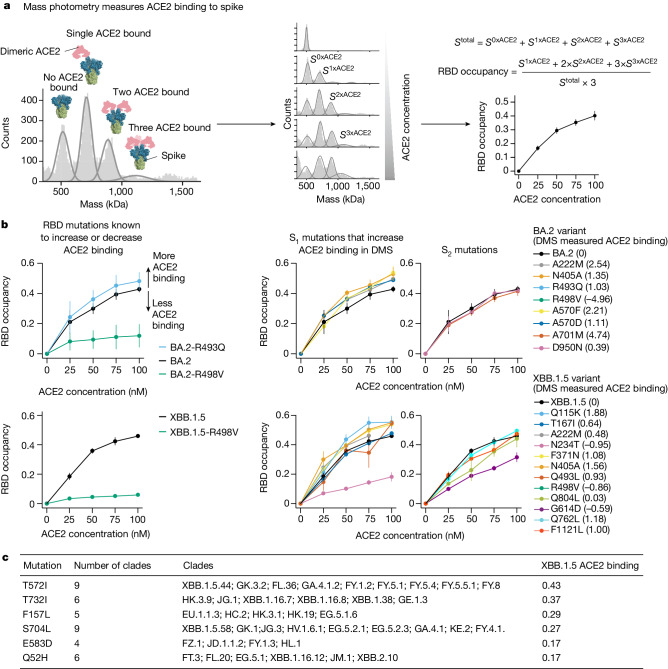
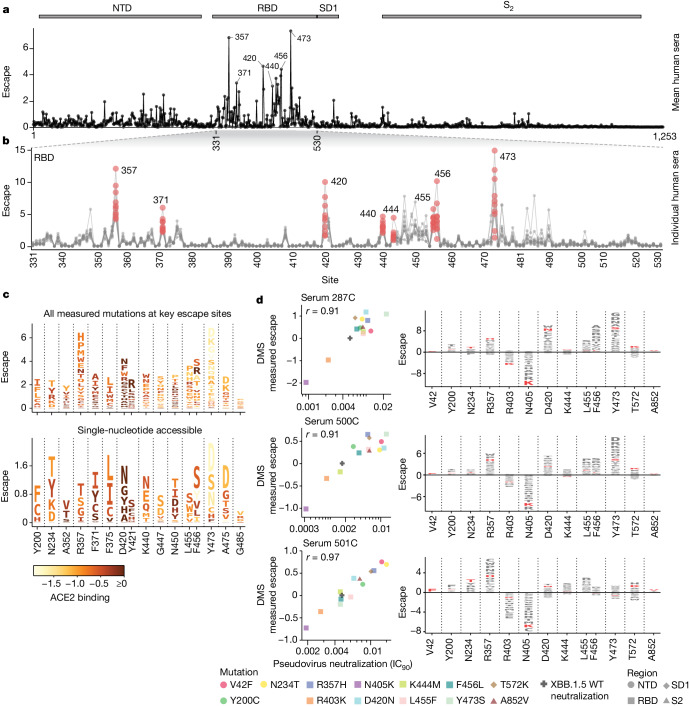
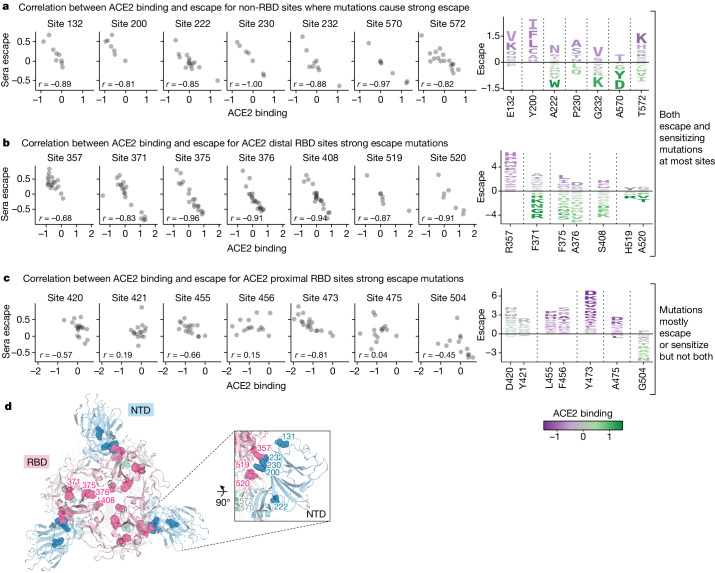
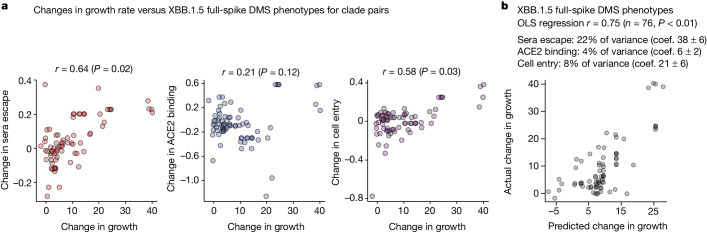
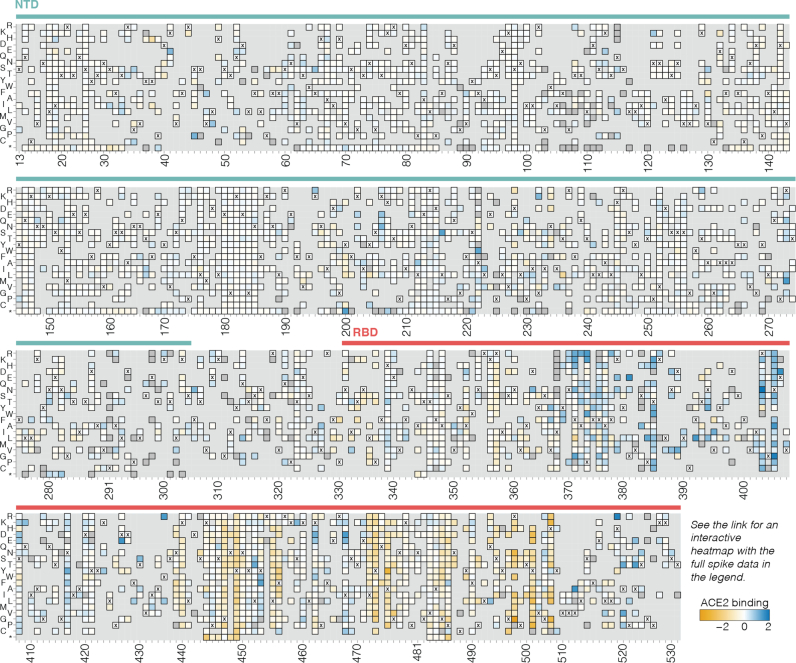
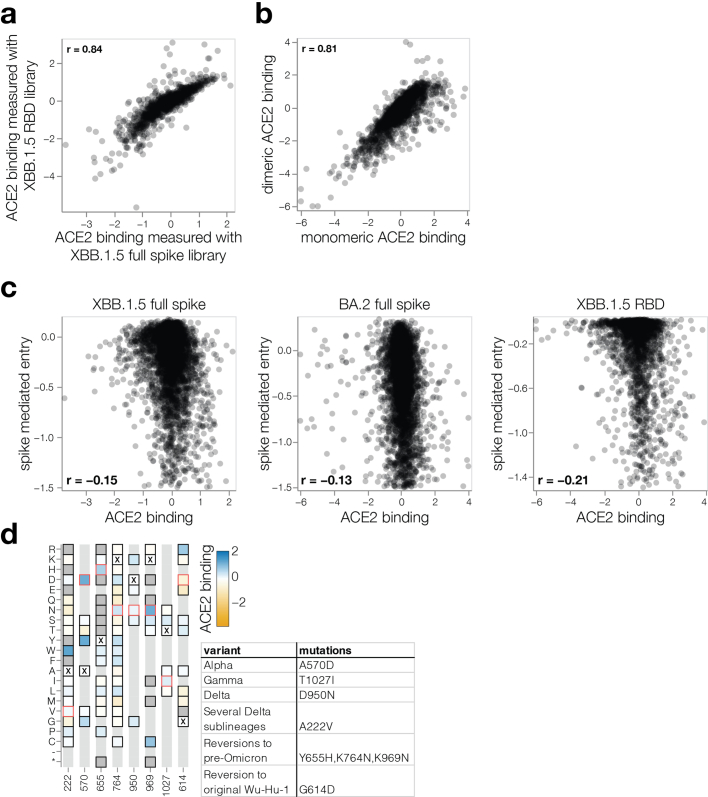
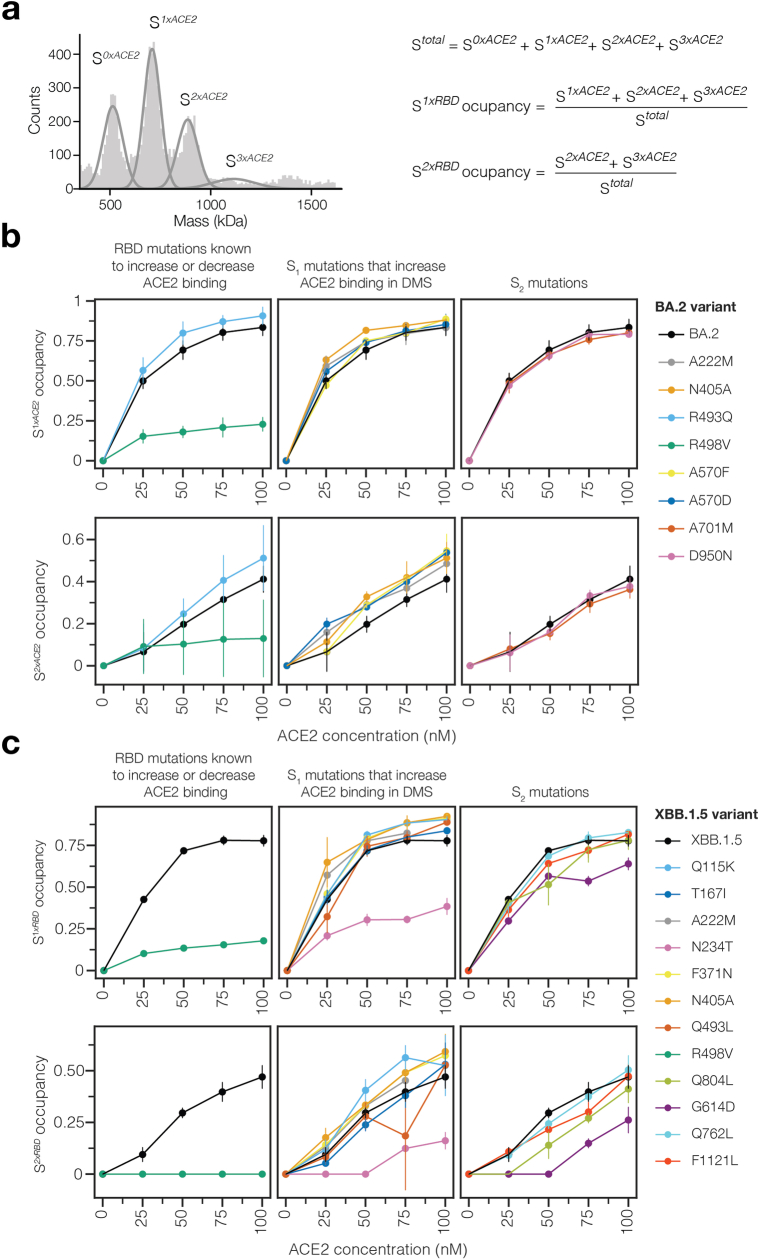
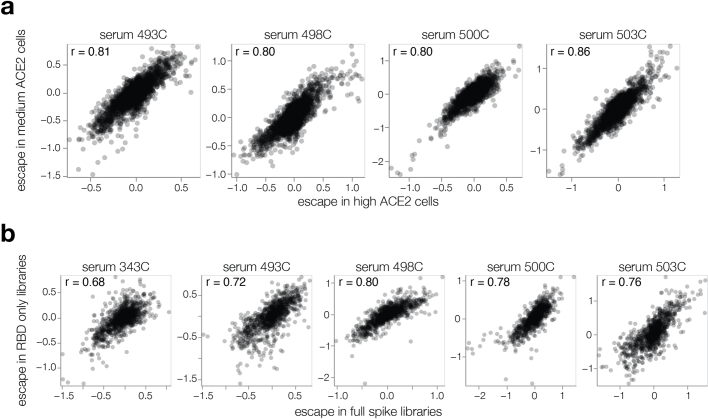
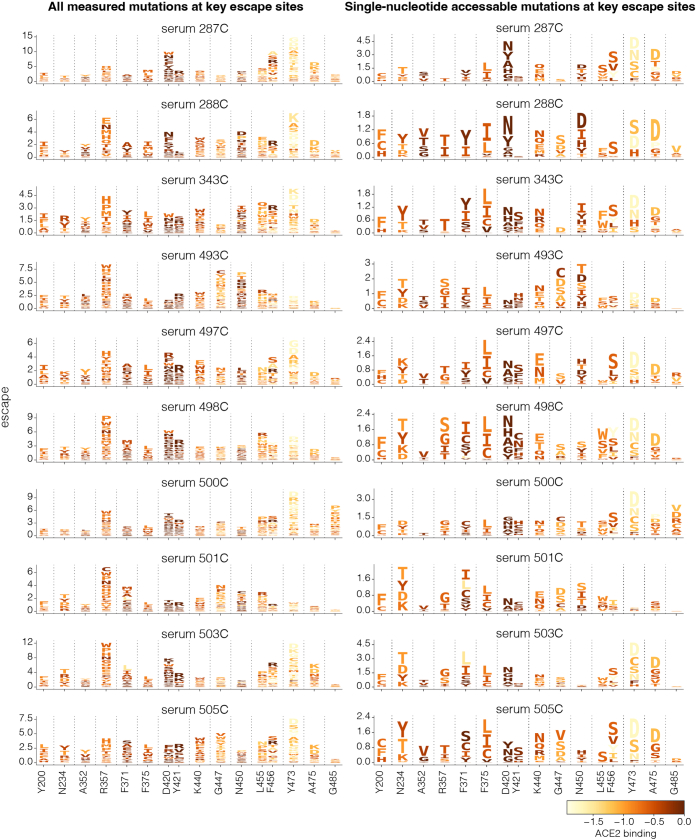
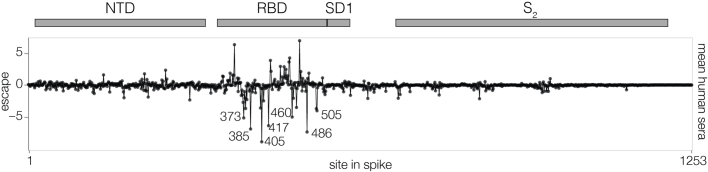
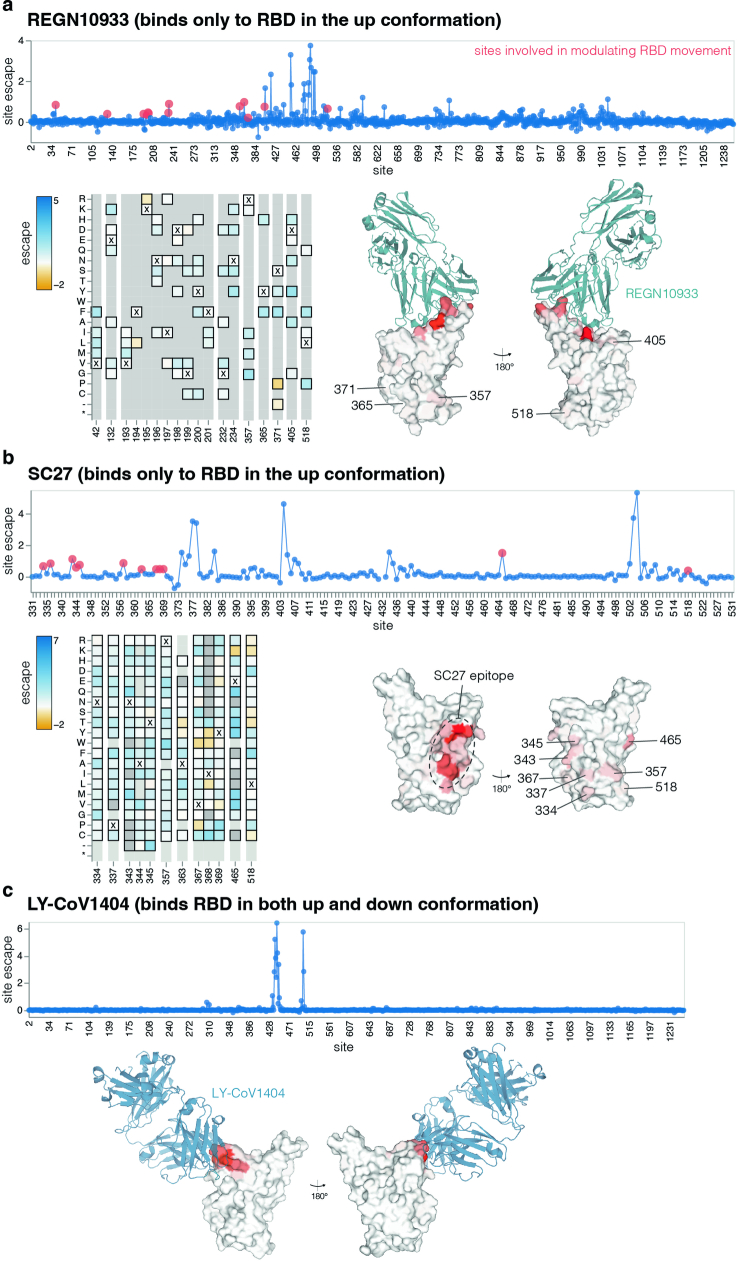
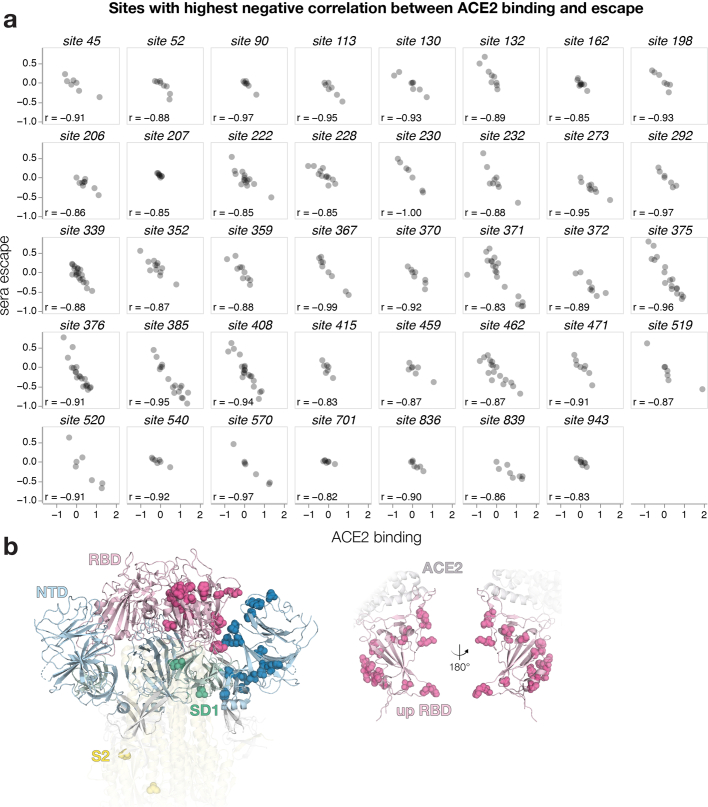
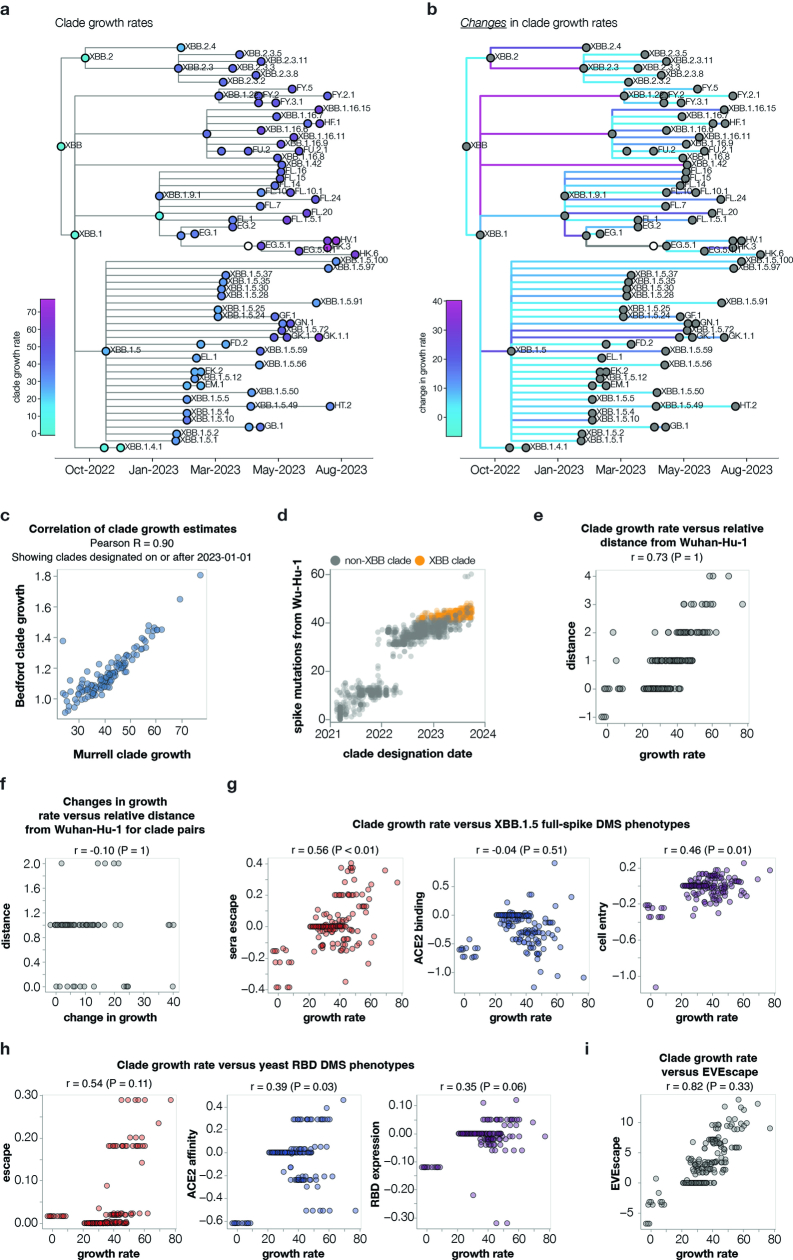
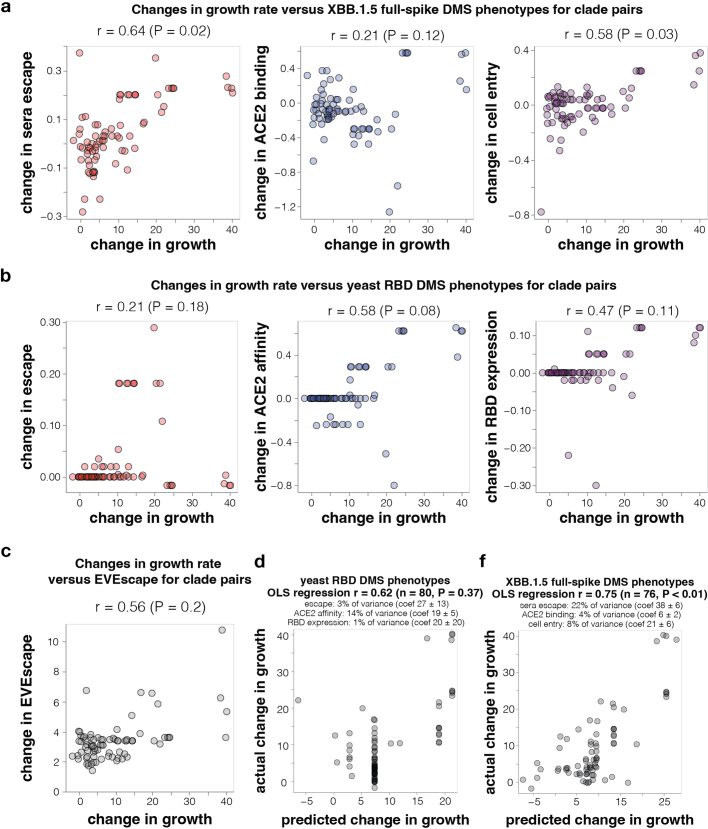
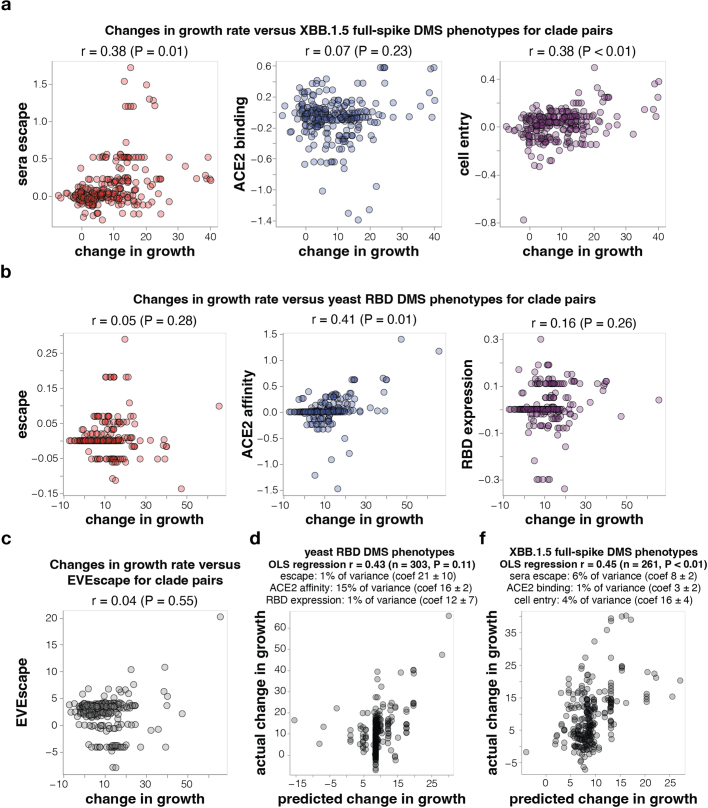
SARS-CoV-2 variants acquire mutations in the spike protein that promote immune evasion1 and affect other properties that contribute to viral fitness, such as ACE2 receptor binding and cell entry2,3. Knowledge of how mutations affect these spike phenotypes can provide insight into the current and potential future evolution of the virus. Here we use pseudovirus deep mutational scanning4 to measure how more than 9,000 mutations across the full XBB.1.5 and BA.2 spikes affect ACE2 binding, cell entry or escape from human sera. We find that mutations outside the receptor-binding domain (RBD) have meaningfully affected ACE2 binding during SARS-CoV-2 evolution. We also measure how mutations to the XBB.1.5 spike affect neutralization by serum from individuals who recently had SARS-CoV-2 infections. The strongest serum escape mutations are in the RBD at sites 357, 420, 440, 456 and 473; however, the antigenic effects of these mutations vary across individuals. We also identify strong escape mutations outside the RBD; however, many of them decrease ACE2 binding, suggesting they act by modulating RBD conformation. Notably, the growth rates of human SARS-CoV-2 clades can be explained in substantial part by the measured effects of mutations on spike phenotypes, suggesting our data could enable better prediction of viral evolution.
© 2024. The Author(s).
Conflict of interest statement
J.D.B. and B.D. are inventors on Fred Hutchinson Cancer Center licensed patents related to the pseudovirus deep mutational scanning system used in this paper. J.D.B. consults for Apriori Bio, Invivyd, Aerium Therapeutics, GlaxoSmithKline and the Vaccine Company on topics related to viral evolution. H.Y.C. reports consulting with Ellume, Pfizer and the Bill and Melinda Gates Foundation. She has served on advisory boards for Vir, Merck and Abbvie. She has conducted continuing medical education teaching with Medscape, Vindico and Clinical Care Options. She has received research funding from Gates Ventures, and support and reagents from Ellume and Cepheid, all outside the submitted work. D.V. is named as inventor on patents for coronavirus vaccines filed by the University of Washington. The other authors declare no competing interests.
Figures
Update of
-
Full-spike deep mutational scanning helps predict the evolutionary success of SARS-CoV-2 clades.bioRxiv [Preprint]. 2023 Nov 14:2023.11.13.566961. doi: 10.1101/2023.11.13.566961. bioRxiv. 2023. Update in: Nature. 2024 Jul;631(8021):617-626. doi: 10.1038/s41586-024-07636-1. PMID: 38014024 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
