

N-acetyltransferase 10 facilitates tumorigenesis of diffuse large B-cell lymphoma by regulating AMPK/mTOR signalling through N4-acetylcytidine modification of SLC30A9
- PMID: 38961519
- PMCID: PMC11222071
- DOI: 10.1002/ctm2.1747
N-acetyltransferase 10 facilitates tumorigenesis of diffuse large B-cell lymphoma by regulating AMPK/mTOR signalling through N4-acetylcytidine modification of SLC30A9
Abstract
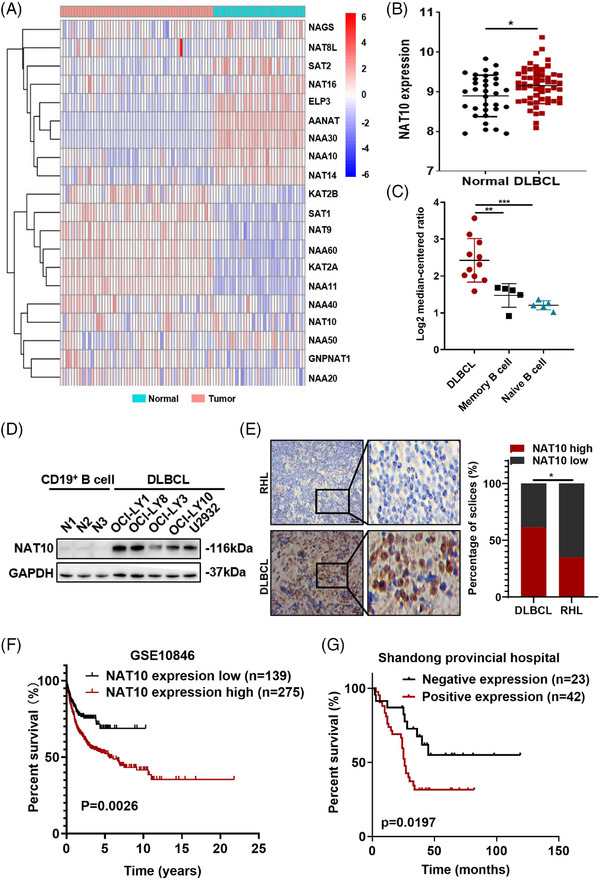
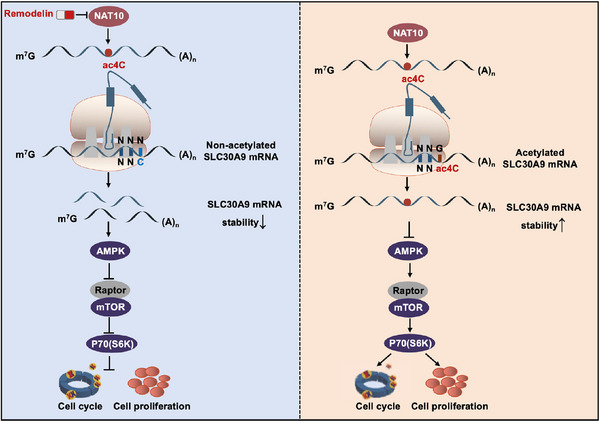
Background: Accumulating studies suggested that posttranscriptional modifications exert a vital role in the tumorigenesis of diffuse large B-cell lymphoma (DLBCL). N4-acetylcytidine (ac4C) modification, catalyzed by the N-acetyltransferase 10 (NAT10), was a novel type of chemical modification that improves translation efficiency and mRNA stability.
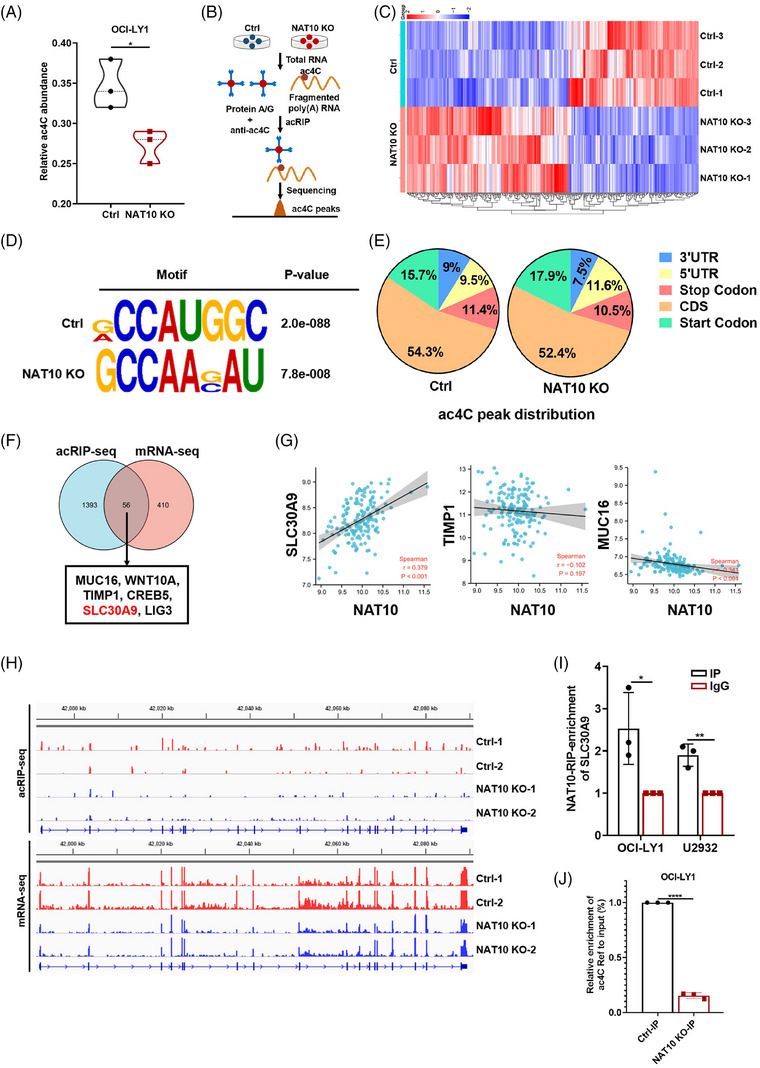
Methods: GEO databases and clinical samples were used to explore the expression and clinical value of NAT10 in DLBCL. CRISPER/Cas9-mediated knockout of NAT10 was performed to determine the biological functions of NAT10 in DLBCL. RNA sequencing, acetylated RNA immunoprecipitation sequencing (acRIP-seq), LC-MS/MS, RNA immunoprecipitation (RIP)-qPCR and RNA stability assays were performed to explore the mechanism by which NAT10 contributed to DLBCL progression.
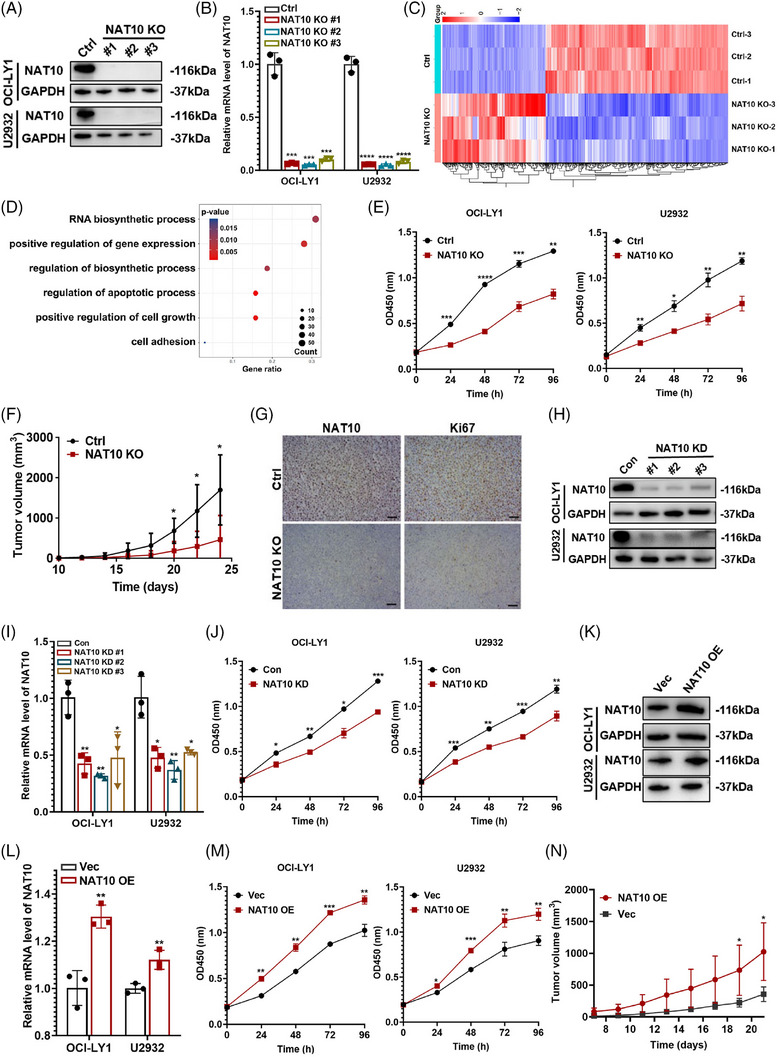
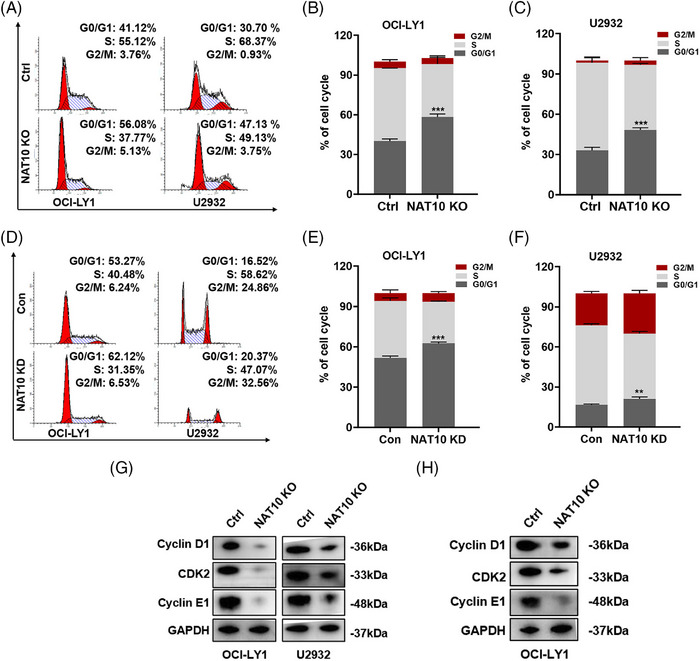
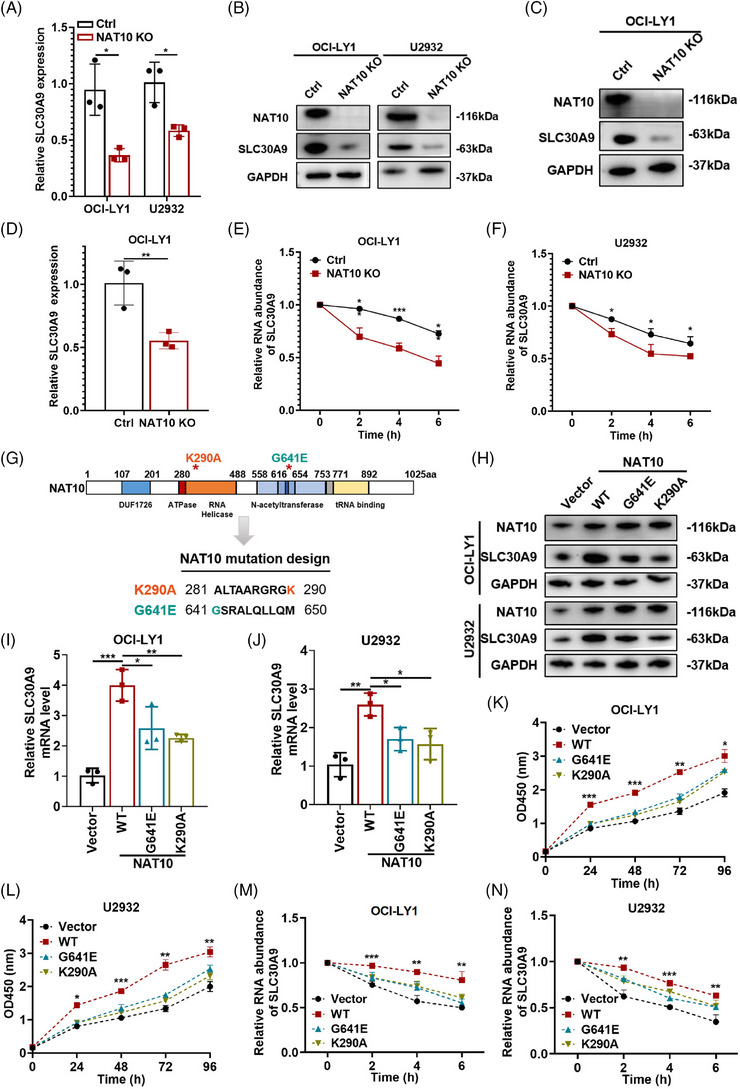
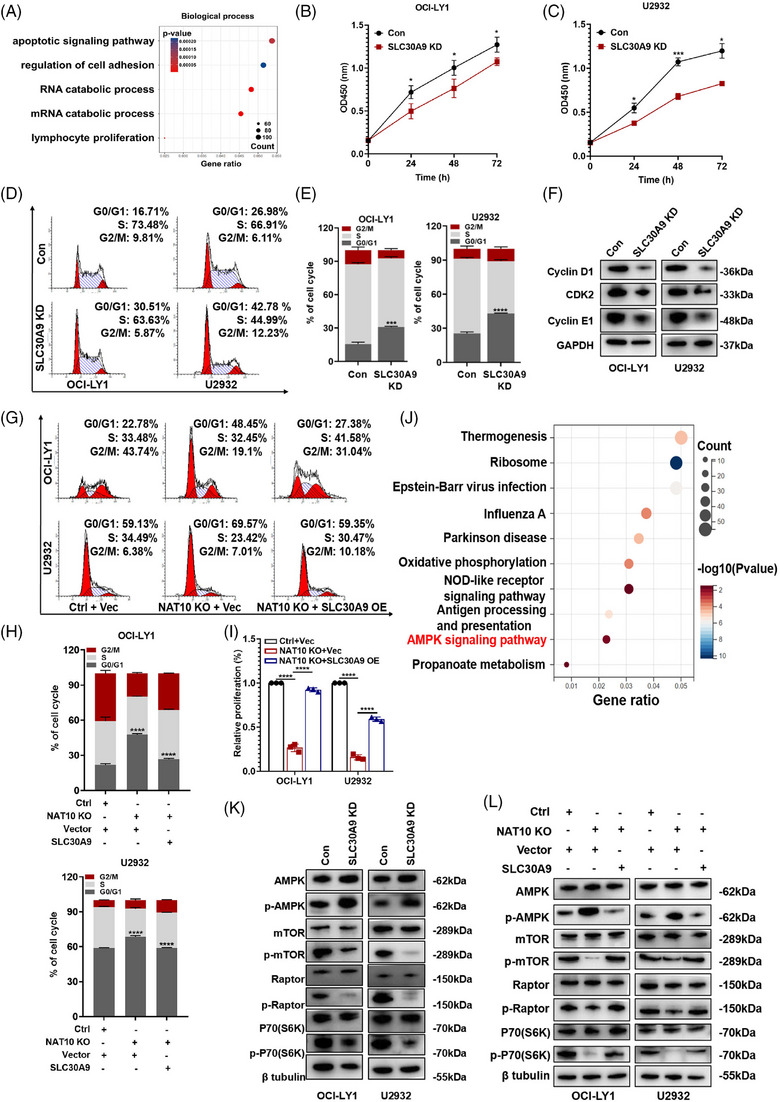
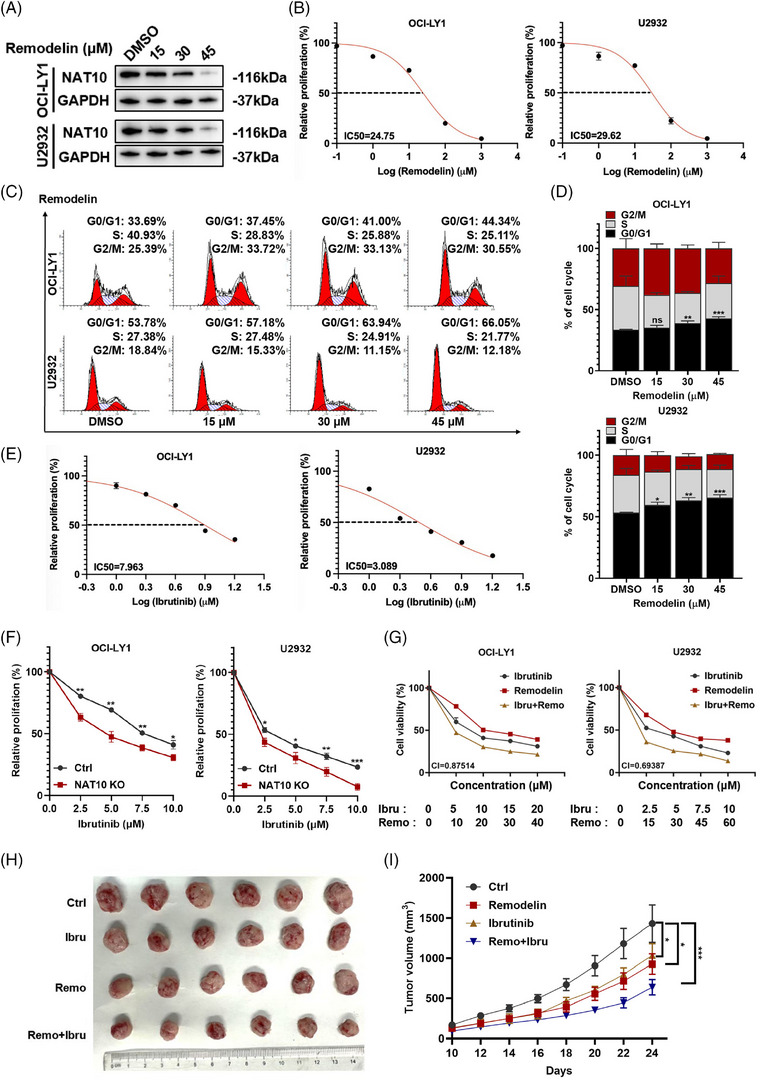
Results: Here, we demonstrated that NAT10-mediated ac4C modification regulated the occurrence and progression of DLBCL. Dysregulated N-acetyltransferases expression was found in DLBCL samples. High expression of NAT10 was associated with poor prognosis of DLBCL patients. Deletion of NAT10 expression inhibited cell proliferation and induced G0/G1 phase arrest. Furthermore, knockout of NAT10 increased the sensitivity of DLBCL cells to ibrutinib. AcRIP-seq identified solute carrier family 30 member 9 (SLC30A9) as a downstream target of NAT10 in DLBCL. NAT10 regulated the mRNA stability of SLC30A9 in an ac4C-dependent manner. Genetic silencing of SLC30A9 suppressed DLBCL cell growth via regulating the activation of AMP-activated protein kinase (AMPK) pathway.
Conclusion: Collectively, these findings highlighted the essential role of ac4C RNA modification mediated by NAT10 in DLBCL, and provided insights into novel epigenetic-based therapeutic strategies.
Keywords: Diffuse large B‐cell lymphoma; N4‐acetyclytidine; N‐acetyltransferase 10; Solute carrier family 30 member 9.
© 2024 The Author(s). Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
Similar articles
-
Emerging Role of NAT10 as ac4C Writer in Inflammatory Diseases: Mechanisms and Therapeutic Applications.Curr Drug Targets. 2025;26(4):282-294. doi: 10.2174/0113894501346709241202110834. Curr Drug Targets. 2025. PMID: 39633518 Review.
-
The role and mechanism of NAT10-mediated ac4C modification in tumor development and progression.MedComm (2020). 2024 Dec 4;5(12):e70026. doi: 10.1002/mco2.70026. eCollection 2024 Dec. MedComm (2020). 2024. PMID: 39640362 Free PMC article. Review.
-
Targeting N4-acetylcytidine suppresses hepatocellular carcinoma progression by repressing eEF2-mediated HMGB2 mRNA translation.Cancer Commun (Lond). 2024 Sep;44(9):1018-1041. doi: 10.1002/cac2.12595. Epub 2024 Jul 19. Cancer Commun (Lond). 2024. PMID: 39030964 Free PMC article.
-
N-acetyltransferase 10 regulates alphavirus replication via N4-acetylcytidine (ac4C) modification of the lymphocyte antigen six family member E (LY6E) mRNA.J Virol. 2024 Jan 23;98(1):e0135023. doi: 10.1128/jvi.01350-23. Epub 2024 Jan 3. J Virol. 2024. PMID: 38169284 Free PMC article.
-
NAT10-mediated mRNA N4-acetylcytidine modification promotes bladder cancer progression.Clin Transl Med. 2022 May;12(5):e738. doi: 10.1002/ctm2.738. Clin Transl Med. 2022. PMID: 35522942 Free PMC article.
Cited by
-
Emerging Role of NAT10 as ac4C Writer in Inflammatory Diseases: Mechanisms and Therapeutic Applications.Curr Drug Targets. 2025;26(4):282-294. doi: 10.2174/0113894501346709241202110834. Curr Drug Targets. 2025. PMID: 39633518 Review.
-
High oxidative phosphorylation represented by UQCRFS1 marks CD8 + tumor-infiltrating lymphocytes exhaustion in diffuse large B-cell lymphoma.Biol Direct. 2025 Aug 13;20(1):92. doi: 10.1186/s13062-025-00684-1. Biol Direct. 2025. PMID: 40804681 Free PMC article.
-
The role and mechanism of NAT10-mediated ac4C modification in tumor development and progression.MedComm (2020). 2024 Dec 4;5(12):e70026. doi: 10.1002/mco2.70026. eCollection 2024 Dec. MedComm (2020). 2024. PMID: 39640362 Free PMC article. Review.
-
N-acetyltransferase 10 as a novel prognostic biomarker in papillary renal cell carcinoma: a machine learning and experimental validation study.Transl Cancer Res. 2024 Nov 30;13(11):6364-6380. doi: 10.21037/tcr-2024-2195. Epub 2024 Nov 27. Transl Cancer Res. 2024. PMID: 39697710 Free PMC article.
-
CRISPR/Cas9 System as a Promising Therapy in Thalassemia and Sickle Cell Disease: A Systematic Review of Clinical Trials.Mol Biotechnol. 2025 Jan 10. doi: 10.1007/s12033-025-01368-x. Online ahead of print. Mol Biotechnol. 2025. PMID: 39794549 Review.
References
MeSH terms
Substances
Grants and funding
- 2018CXGC1213/Key Research and Development Program of Shandong Province
- 2021T1404223/China Postdoctoral Science Foundation
- 2021WWB02/Translational Research Grant of NCRCH
- 2020ZKMB01/Translational Research Grant of NCRCH
- ZR2021YQ51/Shandong Provincial Natural Science Foundation
- tspd20230610/Taishan Scholars Program of Shandong Province
- Shandong Provincial Engineering Research Center of Lymphoma
- 2019QL018/Academic Promotion Programme of Shandong First Medical University
- 82270200/National Natural Science Foundation of China
- 82170189/National Natural Science Foundation of China
- 82070203/National Natural Science Foundation of China
- 81800194/National Natural Science Foundation of China
- 81770210/National Natural Science Foundation of China
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous