

Exploring Splice-Site Mutations in LAMA2-Related Muscular Dystrophies: A Comprehensive Analysis of Genotypic and Phenotypic Patterns
- PMID: 38962616
- PMCID: PMC11221619
- DOI: 10.7759/cureus.61599
Exploring Splice-Site Mutations in LAMA2-Related Muscular Dystrophies: A Comprehensive Analysis of Genotypic and Phenotypic Patterns
Abstract
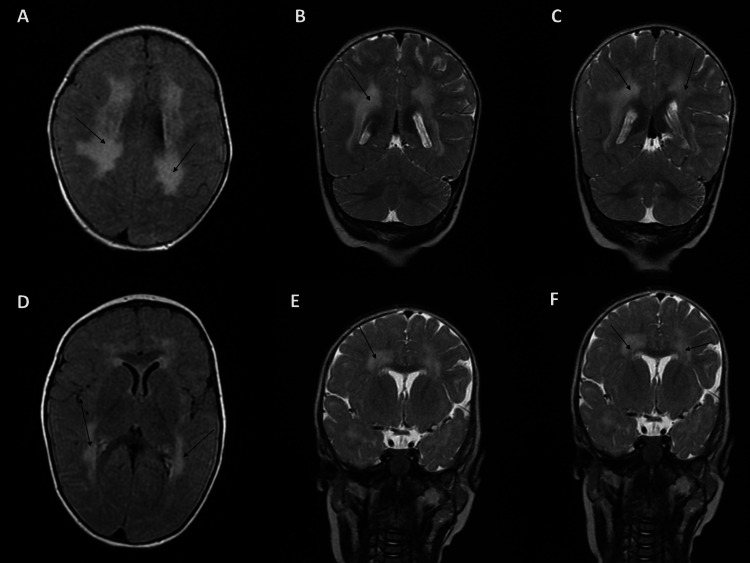
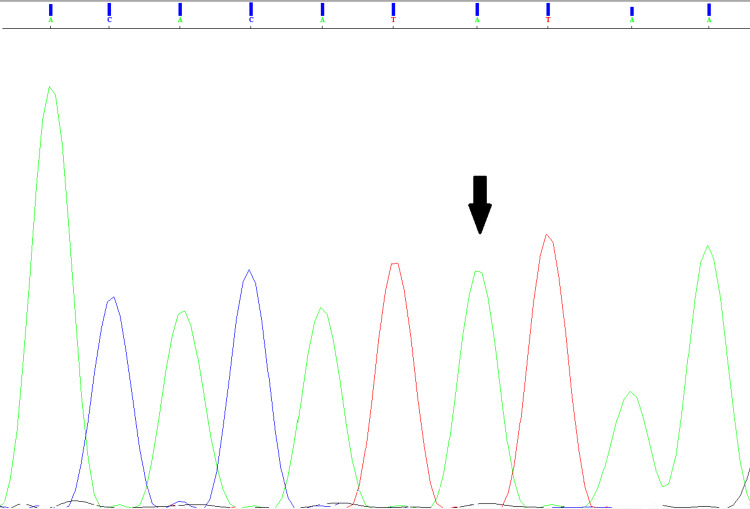
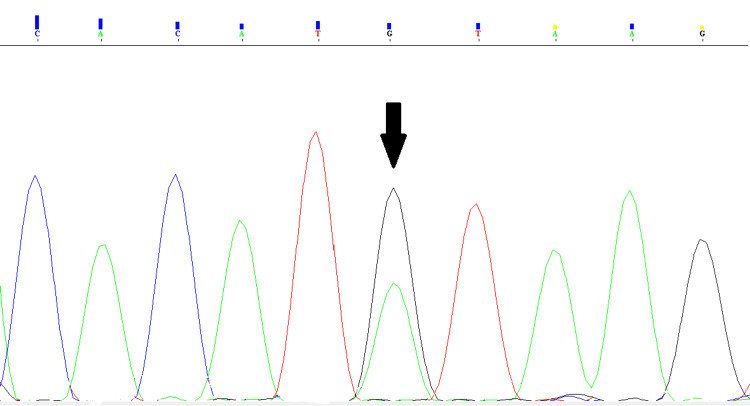
LAMA2-related muscular dystrophies (LAMA2-RDs) constitute the most prevalent subtype of congenital muscular dystrophies (CMDs). The clinical spectrum of LAMA2-RDs exhibits considerable diversity, particularly in motor development and disease progression. Phenotypic variability ranges from severe, early-onset presentation, known as merosin-deficient CMD type 1A, to milder, late-onset presentations, including limb-girdle muscular dystrophy-like phenotype. In this study, whole exome sequencing (WES) was applied to a family with a single proband affected by severe muscular dystrophy. The identified causative mutation was a biallelic splice-site mutation in intron 58 of the LAMA2 gene, leading to a premature termination codon in the critical G domain of laminin-α2 and resulting in a severe phenotype. Additionally, we summarized previously reported splice-site mutations to investigate the clinical and transcription consequences of these mutations. Our findings conclude that splice-site mutations predominantly lead to severe MDC1A, whether in a homozygous or heterozygous state, often associated with another loss-of-function mutation. Besides, splice-site mutations with available analysis of their transcriptional consequences were found to be responsible for exon skipping in most cases and the loss of the reading frame. These findings revealed the importance of WES in identifying disease-causing mutations, particularly in highly diversified pathologies like LAMA2-RDs. The results also underscore the importance of transcriptional analysis in determining the impact of splice-site mutations and the phenotype of LAMA2-RDs on patients.
Keywords: lama2-related muscular dystrophies; laminin-α2; lgmd-like phenotype; mdc1a; splice-site mutations; whole exome sequencing.
Copyright © 2024, Nmer et al.
Conflict of interest statement
Human subjects: Consent was obtained or waived by all participants in this study. Conflicts of interest: In compliance with the ICMJE uniform disclosure form, all authors declare the following: Payment/services info: All authors have declared that no financial support was received from any organization for the submitted work. Financial relationships: All authors have declared that they have no financial relationships at present or within the previous three years with any organizations that might have an interest in the submitted work. Other relationships: All authors have declared that there are no other relationships or activities that could appear to have influenced the submitted work.
Figures
Similar articles
-
Identification of a compound heterozygous missense mutation in LAMA2 gene from a patient with merosin-deficient congenital muscular dystrophy type 1A.J Clin Lab Anal. 2021 Nov;35(11):e23930. doi: 10.1002/jcla.23930. Epub 2021 Sep 16. J Clin Lab Anal. 2021. PMID: 34528292 Free PMC article.
-
Mutations in LAMA2 and CAPN3 genes associated with genetic and phenotypic heterogeneities within a single consanguineous family involving both congenital and progressive muscular dystrophies.Biosci Rep. 2011 Apr;31(2):125-35. doi: 10.1042/BSR20100026. Biosci Rep. 2011. PMID: 20477750
-
A novel de novo variant of LAMA2 contributes to merosin deficient congenital muscular dystrophy type 1A: Case report.Biomed Rep. 2020 Feb;12(2):46-50. doi: 10.3892/br.2019.1260. Epub 2019 Nov 27. Biomed Rep. 2020. PMID: 31929873 Free PMC article.
-
LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness.Front Mol Neurosci. 2020 Aug 5;13:123. doi: 10.3389/fnmol.2020.00123. eCollection 2020. Front Mol Neurosci. 2020. PMID: 32848593 Free PMC article. Review.
-
LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes.Hum Mutat. 2018 Oct;39(10):1314-1337. doi: 10.1002/humu.23599. Epub 2018 Aug 10. Hum Mutat. 2018. PMID: 30055037 Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous