

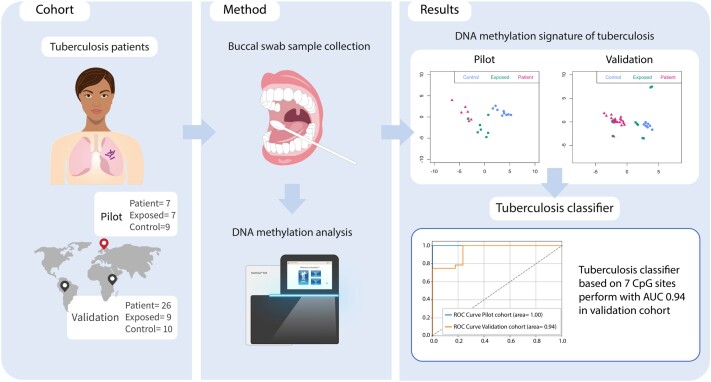
A DNA Methylation Signature From Buccal Swabs to Identify Tuberculosis Infection
- PMID: 38962817
- PMCID: PMC11793033
- DOI: 10.1093/infdis/jiae333
A DNA Methylation Signature From Buccal Swabs to Identify Tuberculosis Infection
Abstract
Background: Tuberculosis (TB) is among the largest infectious causes of death worldwide, and there is a need for a time- and resource-effective diagnostic methods. In this novel and exploratory study, we show the potential of using buccal swabs to collect human DNA and investigate the DNA methylation (DNAm) signatures as a diagnostic tool for TB.
Methods: Buccal swabs were collected from patients with pulmonary TB (n = 7), TB-exposed persons (n = 7), and controls (n = 9) in Sweden. Using Illumina MethylationEPIC array, the DNAm status was determined.
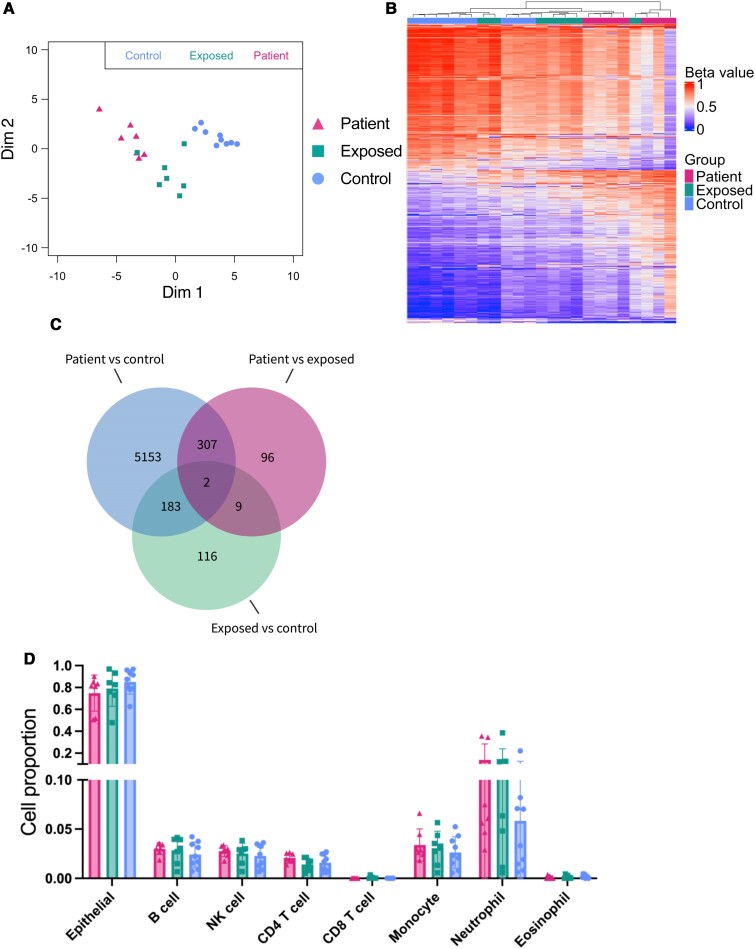
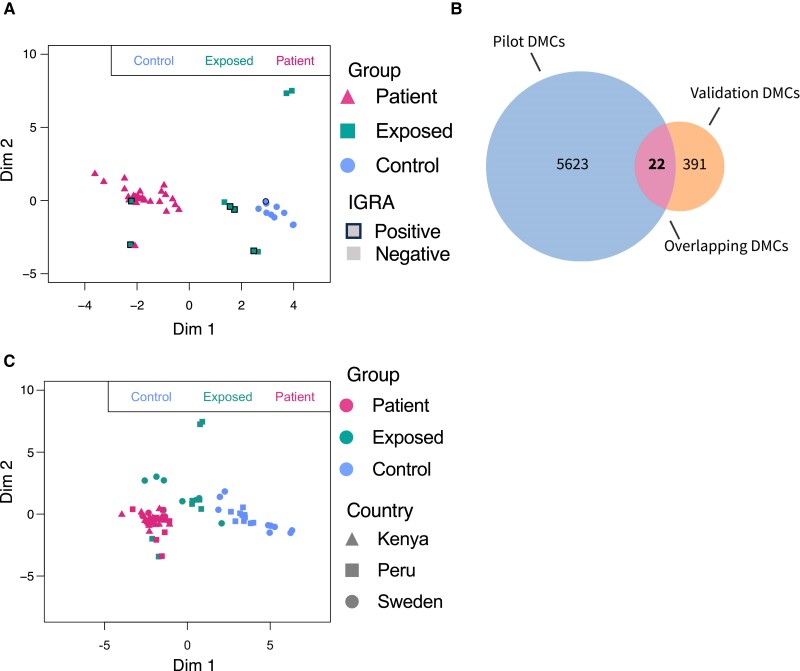
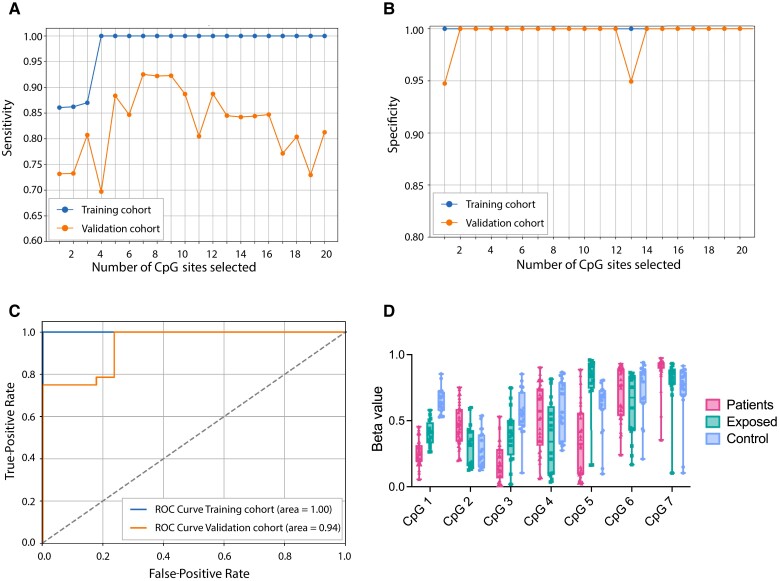
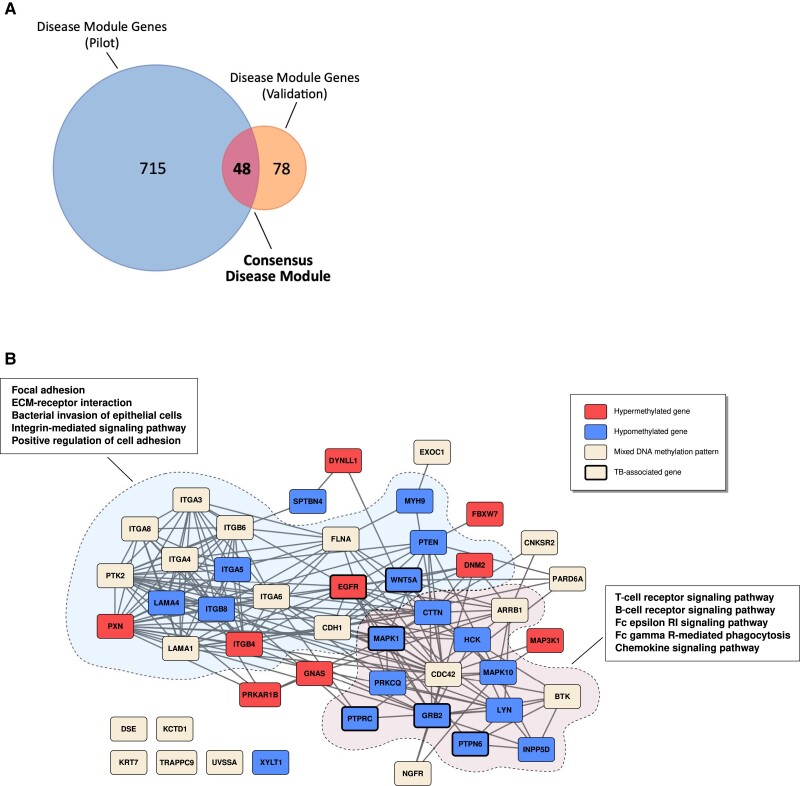
Results: We identified 5644 significant differentially methylated CpG sites between the patients and controls. Performing the analysis on a validation cohort of samples collected in Kenya and Peru (patients, n = 26; exposed, n = 9; control, n = 10) confirmed the DNAm signature. We identified a TB consensus disease module, significantly enriched in TB-associated genes. Last, we used machine learning to identify a panel of 7 CpG sites discriminative for TB and developed a TB classifier. In the validation cohort, the classifier performed with an area under the curve of 0.94, sensitivity of 0.92, and specificity of 1.
Conclusions: In summary, the result from this study shows clinical implications of using DNAm signatures from buccal swabs to explore new diagnostic strategies for TB.
Keywords: DNA methylation; biosignature; buccal swabs; classifier; tuberculosis.
© The Author(s) 2024. Published by Oxford University Press on behalf of Infectious Diseases Society of America.
Conflict of interest statement
Potential conflicts of interest. M. L. and M. G. are founders of PredictME AB. S. S. and D. M.-E. are bioinformaticians at PredictME. All other authors report no potential conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Figures
References
-
- World Health Organization . Global tuberculosis report. 2023. https://www.who.int/publications/i/item/9789240083851. Accessed 28 February 2024.
-
- World Health Organization . Implementing the end TB strategy: the essentials 2015. https://iris.who.int/handle/10665/206499. Accessed 8 August 2023.
-
- Cobelens FG, Menzies D, Farhat M. False-positive tuberculin reactions due to non-tuberculous mycobacterial infections. Int J Tuberc Lung Dis 2007; 11:934–5; author reply 5. - PubMed
