

SNORD3A Regulates STING Transcription to Promote Ferroptosis in Acute Kidney Injury
- PMID: 38962954
- PMCID: PMC11434033
- DOI: 10.1002/advs.202400305
SNORD3A Regulates STING Transcription to Promote Ferroptosis in Acute Kidney Injury
Abstract
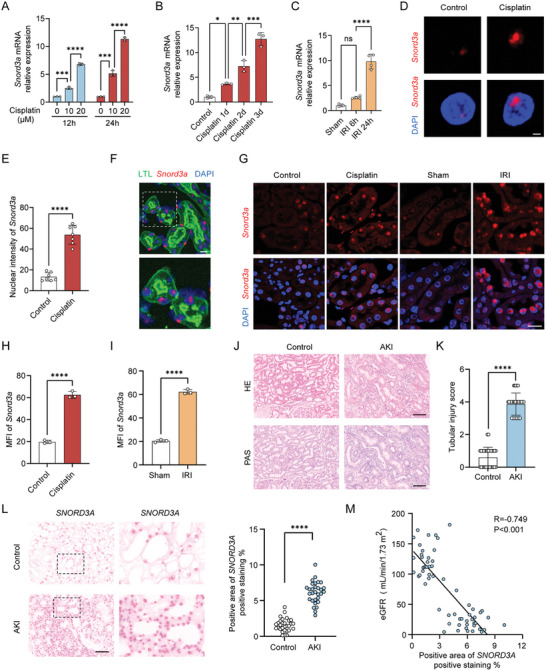
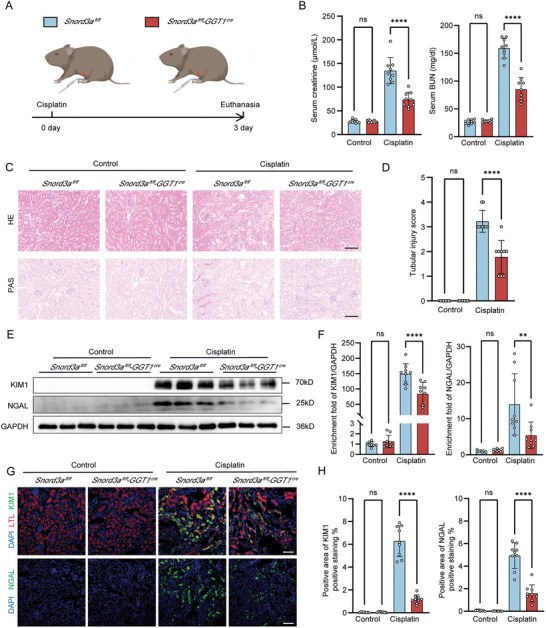
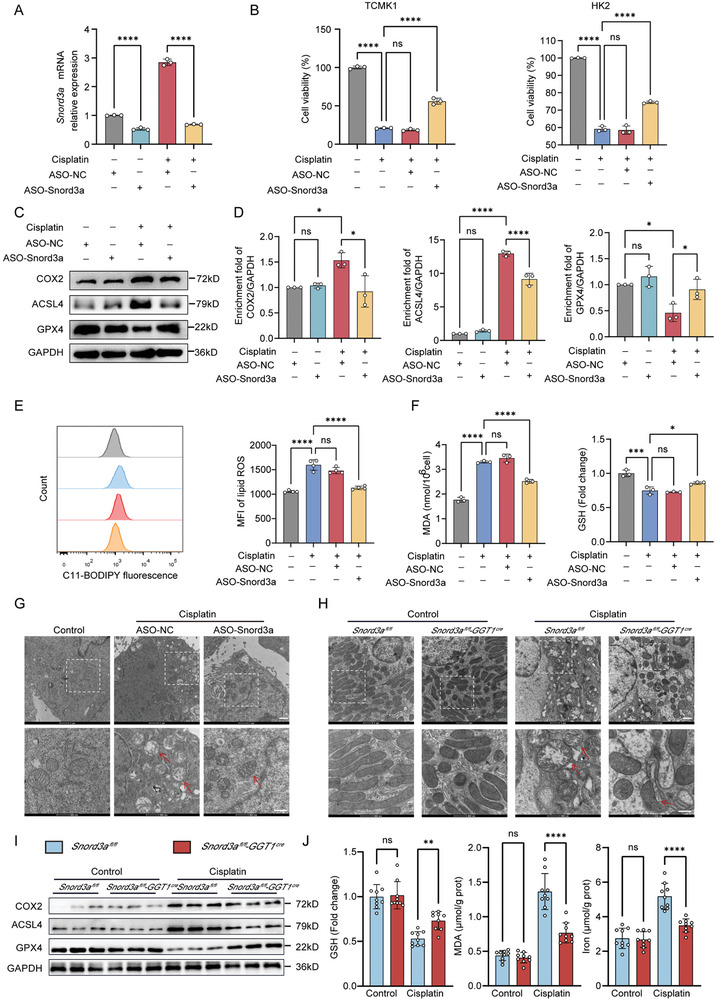
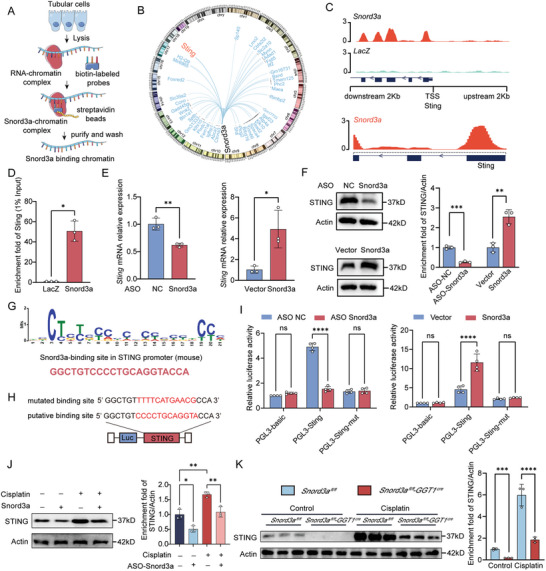
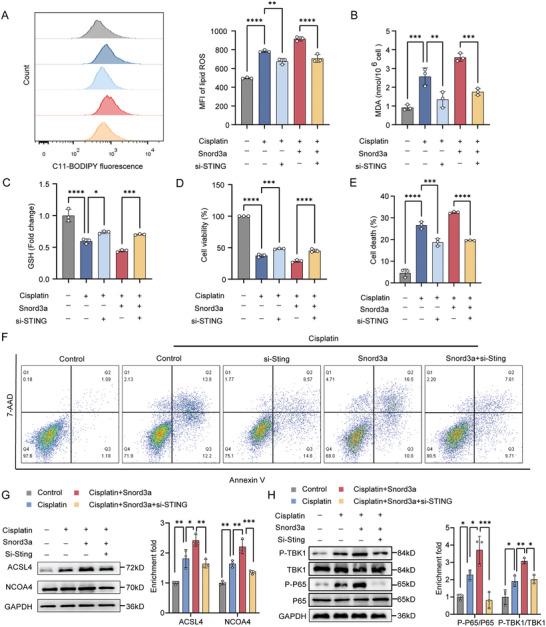
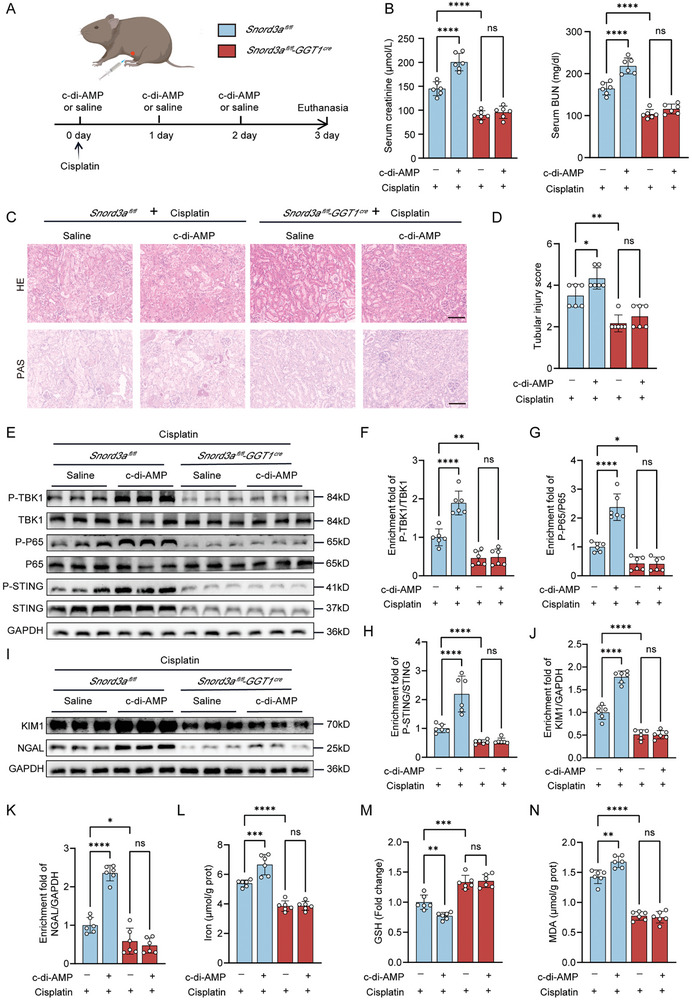
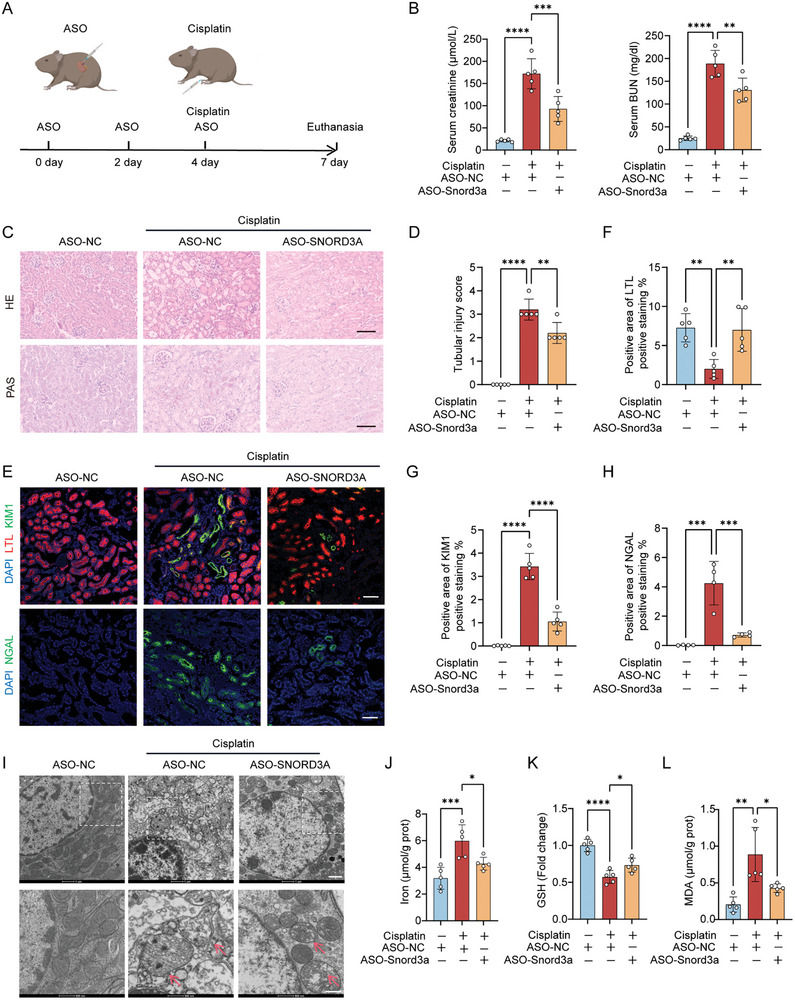
Acute kidney injury (AKI) signifies a sudden and prolonged decline in kidney function characterized by tubular cell death and interstitial inflammation. Small nucleolar RNAs (snoRNAs) play pivotal roles in oxidative stress and inflammation, and may play an important role in the AKI process, which remains elusive. an elevated expression of Snord3a is revealed in renal tubules in response to AKI and demonstrates that Snord3a deficiency alleviates renal injury in AKI mouse models. Notably, the deficiency of Snord3a exhibits a mitigating effect on the stimulator of interferon genes (STING)-associated ferroptosis phenotypes and the progression of tubular injury. Mechanistically, Snord3a is shown to regulate the STING signaling axis via promoting STING gene transcription; administration of Snord3a antisense oligonucleotides establishes a significant therapeutic advantage in AKI mouse models. Together, the findings elucidate the transcription regulation mechanism of STING and the crucial roles of the Snord3a-STING axis in ferroptosis during AKI, underscoring Snord3a as a potential prognostic and therapeutic target for AKI.
Keywords: STING; Snord3a; acute kidney injury; ferroptosis; snoRNA.
© 2024 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- a) Kashani K. B., Awdishu L., Bagshaw S. M., Barreto E. F., Claure‐Del Granado R., Evans B. J., Forni L. G., Ghosh E., Goldstein S. L., Kane‐Gill S. L., Koola J., Koyner J. L., Liu M., Murugan R., Nadkarni G. N., Neyra J. A., Ninan J., Ostermann M., Pannu N., Rashidi P., Ronco C., Rosner M. H., Selby N. M., Shickel B., Singh K., Soranno D. E., Sutherland S. M., Bihorac A., Mehta R. L., Nat. Rev. Nephrol. 2023, 19, 807;
- b) Ronco C., Bellomo R., Kellum J. A., Lancet 2019, 394, 1949. - PubMed
-
- a) Bergeron D., Faucher‐Giguère L., Emmerichs A.‐K., Choquet K., Song K. S., Deschamps‐Francoeur G., Fafard‐Couture É., Rivera A., Couture S., Churchman L. S., Heyd F., Abou Elela S., Scott M. S., Genome Biol. 2023, 24, 160; - PMC - PubMed
- b) Kiss‐László Z., Henry Y., Bachellerie J. P., Caizergues‐Ferrer M., Kiss T., Cell 1996, 85, 1077; - PubMed
- c) Bratkovič T., Božič J., Rogelj B., Nucleic Acids Res. 2020, 48, 1627,. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials