

mLiftOver: harmonizing data across Infinium DNA methylation platforms
- PMID: 38963309
- PMCID: PMC11233119
- DOI: 10.1093/bioinformatics/btae423
mLiftOver: harmonizing data across Infinium DNA methylation platforms
Abstract
Motivation: Infinium DNA methylation BeadChips are widely used for genome-wide DNA methylation profiling at the population scale. Recent updates to probe content and naming conventions in the EPIC version 2 (EPICv2) arrays have complicated integrating new data with previous Infinium array platforms, such as the MethylationEPIC (EPIC) and the HumanMethylation450 (HM450) BeadChip.
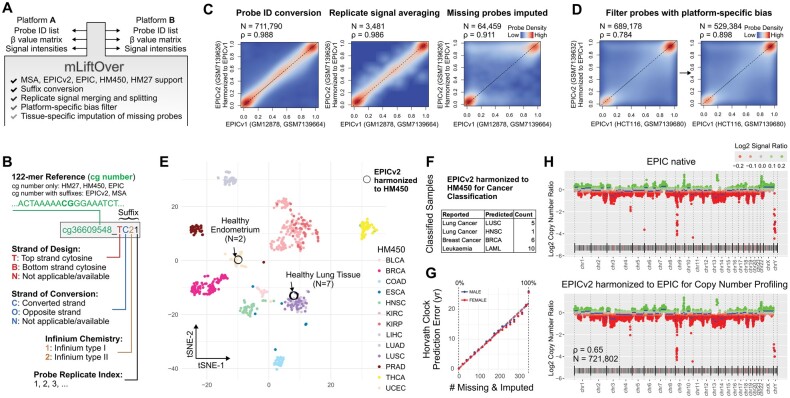
Results: We present mLiftOver, a user-friendly tool that harmonizes probe ID, methylation level, and signal intensity data across different Infinium platforms. It manages probe replicates, missing data imputation, and platform-specific bias for accurate data conversion. We validated the tool by applying HM450-based cancer classifiers to EPICv2 cancer data, achieving high accuracy. Additionally, we successfully integrated EPICv2 healthy tissue data with legacy HM450 data for tissue identity analysis and produced consistent copy number profiles in cancer cells.
Availability and implementation: mLiftOver is implemented R and available in the Bioconductor package SeSAMe (version 1.21.13+): https://bioconductor.org/packages/release/bioc/html/sesame.html. Analysis of EPIC and EPICv2 platform-specific bias and high-confidence mapping is available at https://github.com/zhou-lab/InfiniumAnnotationV1/raw/main/Anno/EPICv2/EPICv2ToEPIC_conversion.tsv.gz. The source code is available at https://github.com/zwdzwd/sesame/blob/devel/R/mLiftOver.R under the MIT license.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
W.Z. received BeadChips from Illumina Inc. for research.
Figures
Similar articles
-
Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling.Genome Biol. 2016 Oct 7;17(1):208. doi: 10.1186/s13059-016-1066-1. Genome Biol. 2016. PMID: 27717381 Free PMC article.
-
Using high-density DNA methylation arrays to profile copy number alterations.Genome Biol. 2014 Feb 3;15(2):R30. doi: 10.1186/gb-2014-15-2-r30. Genome Biol. 2014. PMID: 24490765 Free PMC article.
-
Characterisation and reproducibility of the HumanMethylationEPIC v2.0 BeadChip for DNA methylation profiling.BMC Genomics. 2024 Mar 6;25(1):251. doi: 10.1186/s12864-024-10027-5. BMC Genomics. 2024. PMID: 38448820 Free PMC article.
-
DNA methylation data analysis and its application to cancer research.Epigenomics. 2013 Jun;5(3):301-16. doi: 10.2217/epi.13.26. Epigenomics. 2013. PMID: 23750645 Free PMC article. Review.
-
Review of processing and analysis methods for DNA methylation array data.Br J Cancer. 2013 Sep 17;109(6):1394-402. doi: 10.1038/bjc.2013.496. Epub 2013 Aug 27. Br J Cancer. 2013. PMID: 23982603 Free PMC article. Review.
Cited by
-
Accounting for differences between Infinium MethylationEPIC v2 and v1 in DNA methylation-based tools.Life Sci Alliance. 2025 Jul 8;8(9):e202403155. doi: 10.26508/lsa.202403155. Print 2025 Sep. Life Sci Alliance. 2025. PMID: 40628445 Free PMC article.
-
Accounting for differences between Infinium MethylationEPIC v2 and v1 in DNA methylation-based tools.bioRxiv [Preprint]. 2025 Jun 12:2024.07.02.600461. doi: 10.1101/2024.07.02.600461. bioRxiv. 2025. Update in: Life Sci Alliance. 2025 Jul 8;8(9):e202403155. doi: 10.26508/lsa.202403155. PMID: 39005299 Free PMC article. Updated. Preprint.
References
-
- Bibikova M, Le J, Barnes B. et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 2009;1:177–200. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
