

Unsupervised evolution of protein and antibody complexes with a structure-informed language model
- PMID: 38963838
- PMCID: PMC11616794
- DOI: 10.1126/science.adk8946
Unsupervised evolution of protein and antibody complexes with a structure-informed language model
Abstract
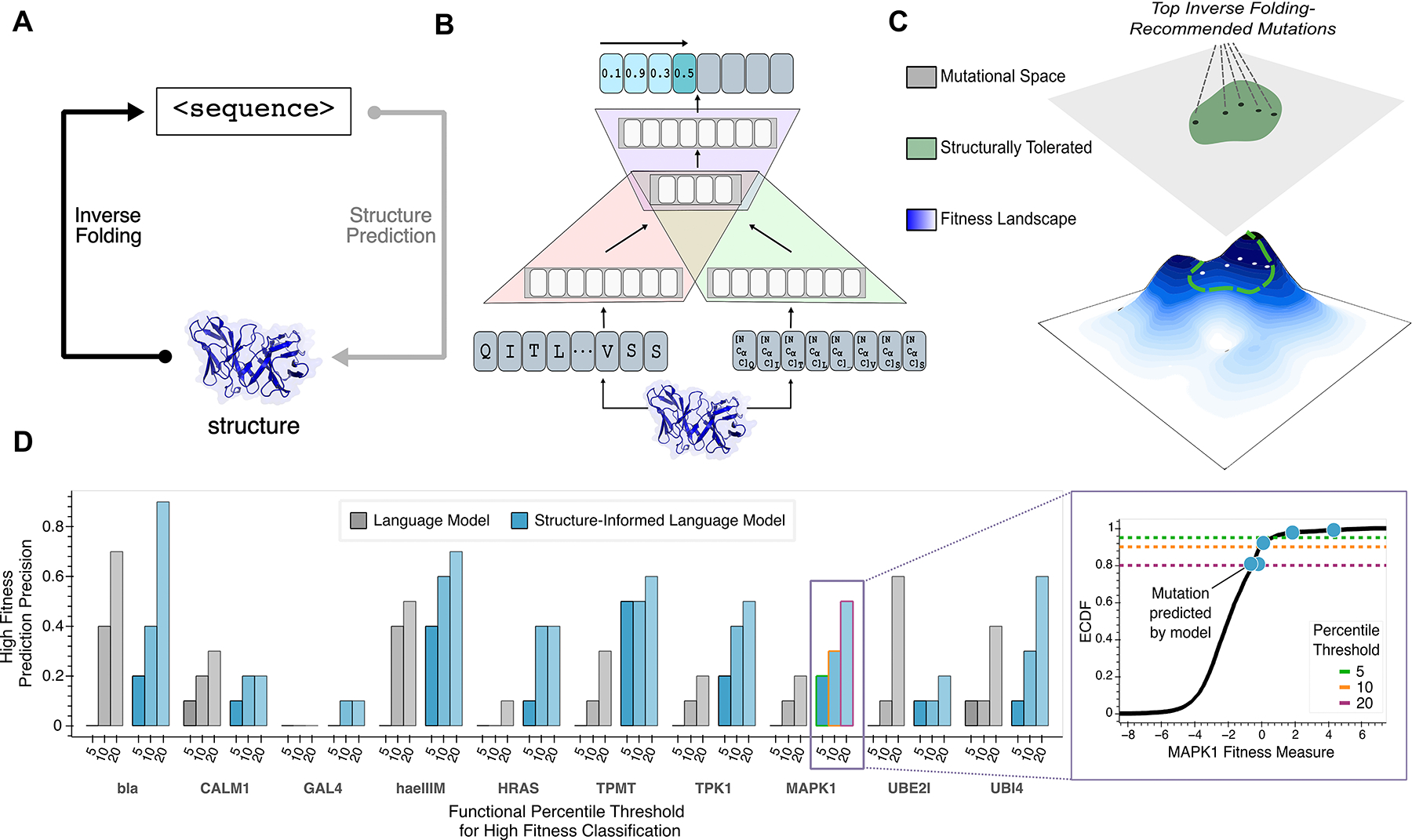
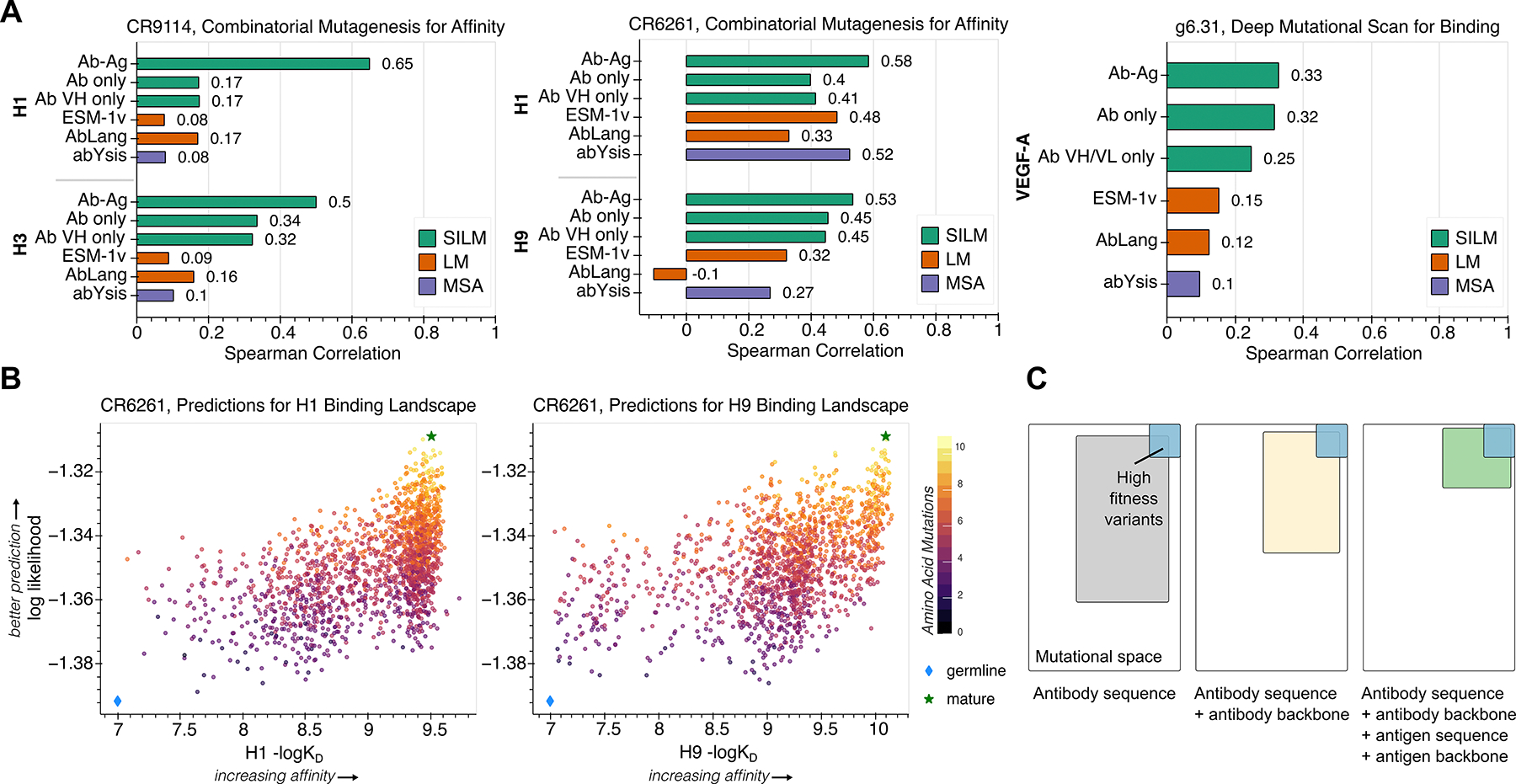
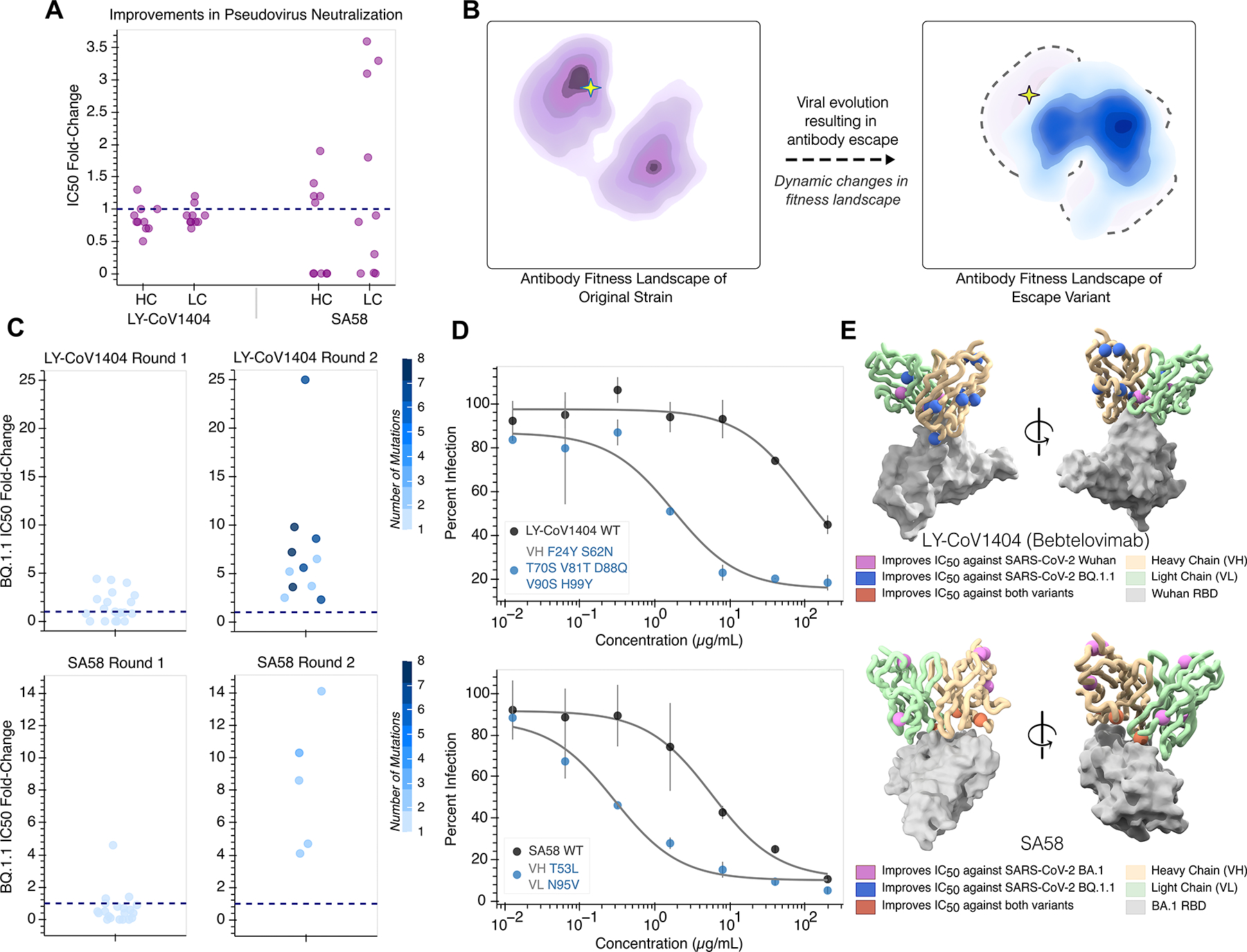
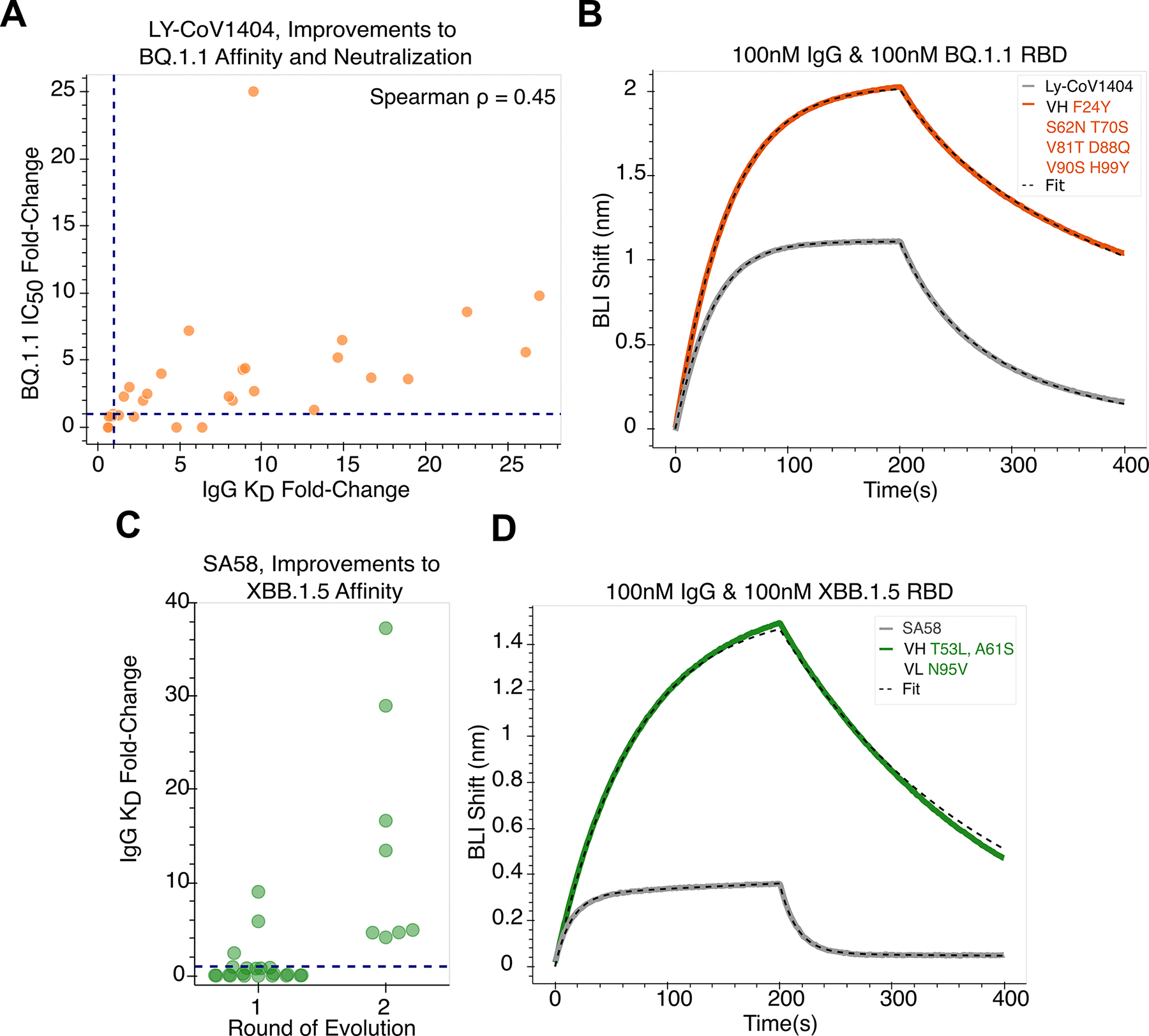
Large language models trained on sequence information alone can learn high-level principles of protein design. However, beyond sequence, the three-dimensional structures of proteins determine their specific function, activity, and evolvability. Here, we show that a general protein language model augmented with protein structure backbone coordinates can guide evolution for diverse proteins without the need to model individual functional tasks. We also demonstrate that ESM-IF1, which was only trained on single-chain structures, can be extended to engineer protein complexes. Using this approach, we screened about 30 variants of two therapeutic clinical antibodies used to treat severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. We achieved up to 25-fold improvement in neutralization and 37-fold improvement in affinity against antibody-escaped viral variants of concern BQ.1.1 and XBB.1.5, respectively. These findings highlight the advantage of integrating structural information to identify efficient protein evolution trajectories without requiring any task-specific training data.
Conflict of interest statement
Competing interests
V.R.S., B.L.H., and P.S.K. are named as inventors on a patent application applied for by Stanford University and the Chan Zuckerberg Biohub entitled “Antibody Compositions and Optimization Methods”. B.L.H acknowledges outside interest in Prox Biosciences as a scientific co-founder.
Figures
Update of
-
Inverse folding of protein complexes with a structure-informed language model enables unsupervised antibody evolution.bioRxiv [Preprint]. 2023 Dec 21:2023.12.19.572475. doi: 10.1101/2023.12.19.572475. bioRxiv. 2023. Update in: Science. 2024 Jul 5;385(6704):46-53. doi: 10.1126/science.adk8946. PMID: 38187780 Free PMC article. Updated. Preprint.
Comment in
-
AI reverse-engineers antibodies.Nat Rev Drug Discov. 2024 Sep;23(9):659. doi: 10.1038/d41573-024-00124-1. Nat Rev Drug Discov. 2024. PMID: 39043932 No abstract available.
Similar articles
-
Inverse folding of protein complexes with a structure-informed language model enables unsupervised antibody evolution.bioRxiv [Preprint]. 2023 Dec 21:2023.12.19.572475. doi: 10.1101/2023.12.19.572475. bioRxiv. 2023. Update in: Science. 2024 Jul 5;385(6704):46-53. doi: 10.1126/science.adk8946. PMID: 38187780 Free PMC article. Updated. Preprint.
-
Affinity maturation of SARS-CoV-2 neutralizing antibodies confers potency, breadth, and resilience to viral escape mutations.Immunity. 2021 Aug 10;54(8):1853-1868.e7. doi: 10.1016/j.immuni.2021.07.008. Epub 2021 Jul 30. Immunity. 2021. PMID: 34331873 Free PMC article.
-
Characterization of MW06, a human monoclonal antibody with cross-neutralization activity against both SARS-CoV-2 and SARS-CoV.MAbs. 2021 Jan-Dec;13(1):1953683. doi: 10.1080/19420862.2021.1953683. MAbs. 2021. PMID: 34313527 Free PMC article.
-
Epitope Analysis of Anti-SARS-CoV-2 Neutralizing Antibodies.Curr Med Sci. 2021 Dec;41(6):1065-1074. doi: 10.1007/s11596-021-2453-8. Epub 2021 Oct 4. Curr Med Sci. 2021. PMID: 34606064 Free PMC article. Review.
-
Analysis of the molecular mechanism of SARS-CoV-2 antibodies.Biochem Biophys Res Commun. 2021 Aug 20;566:45-52. doi: 10.1016/j.bbrc.2021.06.001. Epub 2021 Jun 5. Biochem Biophys Res Commun. 2021. PMID: 34116356 Free PMC article. Review.
Cited by
-
Protein A-like Peptide Design Based on Diffusion and ESM2 Models.Molecules. 2024 Oct 21;29(20):4965. doi: 10.3390/molecules29204965. Molecules. 2024. PMID: 39459333 Free PMC article.
-
De Novo Design of Large Polypeptides Using a Lightweight Diffusion Model Integrating LSTM and Attention Mechanism Under Per-Residue Secondary Structure Constraints.Molecules. 2025 Feb 28;30(5):1116. doi: 10.3390/molecules30051116. Molecules. 2025. PMID: 40076339 Free PMC article.
-
AlphaBind, a domain-specific model to predict and optimize antibody-antigen binding affinity.MAbs. 2025 Dec;17(1):2534626. doi: 10.1080/19420862.2025.2534626. Epub 2025 Jul 22. MAbs. 2025. PMID: 40693434 Free PMC article.
-
Leveraging large language models to predict antibody biological activity against influenza A hemagglutinin.Comput Struct Biotechnol J. 2025 Mar 24;27:1286-1295. doi: 10.1016/j.csbj.2025.03.038. eCollection 2025. Comput Struct Biotechnol J. 2025. PMID: 40230408 Free PMC article.
-
The Nobel Prize in Chemistry: past, present, and future of AI in biology.Commun Biol. 2024 Oct 29;7(1):1409. doi: 10.1038/s42003-024-07113-5. Commun Biol. 2024. PMID: 39472680 Free PMC article.
References
-
- Axe DD, Foster NW, Fersht AR, A Search for Single Substitutions That Eliminate Enzymatic Function in a Bacterial Ribonuclease. Biochemistry 37, 7157–7166 (1998). - PubMed
-
- Shafikhani S, Siegel RA, Ferrari E, Schellenberger V, Generation of large libraries of random mutants in Bacillus subtilis by PCR-based plasmid multimerization. BioTechniques 23, 304–310 (1997). - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
