

Principled distillation of UK Biobank phenotype data reveals underlying structure in human variation
- PMID: 38965376
- PMCID: PMC11343713
- DOI: 10.1038/s41562-024-01909-5
Principled distillation of UK Biobank phenotype data reveals underlying structure in human variation
Abstract
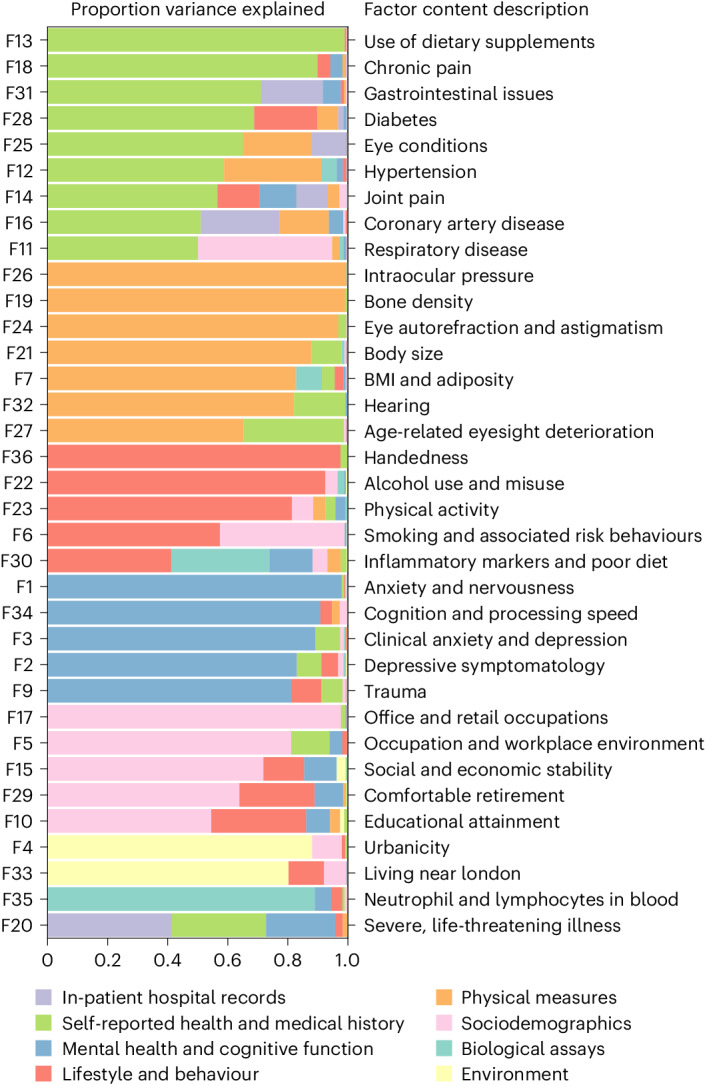
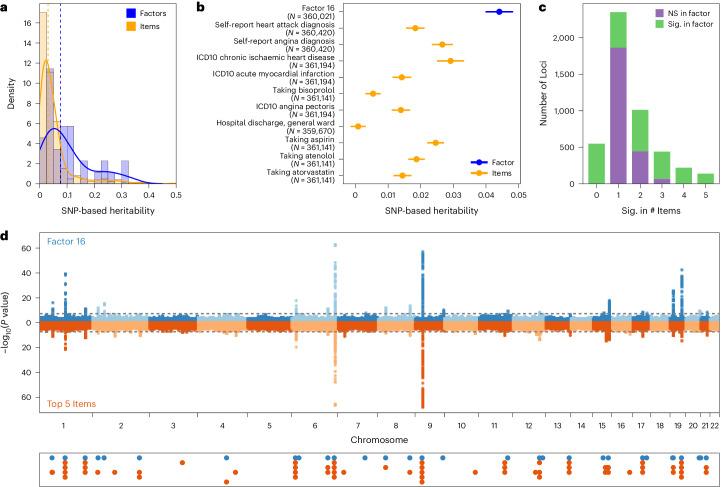
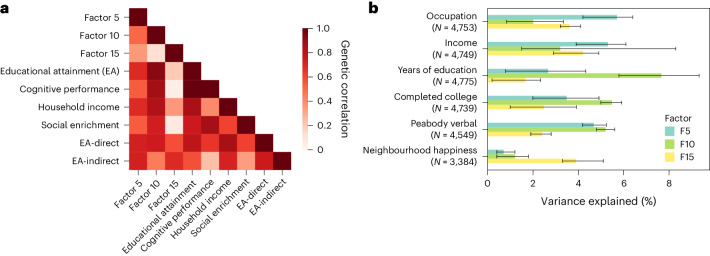
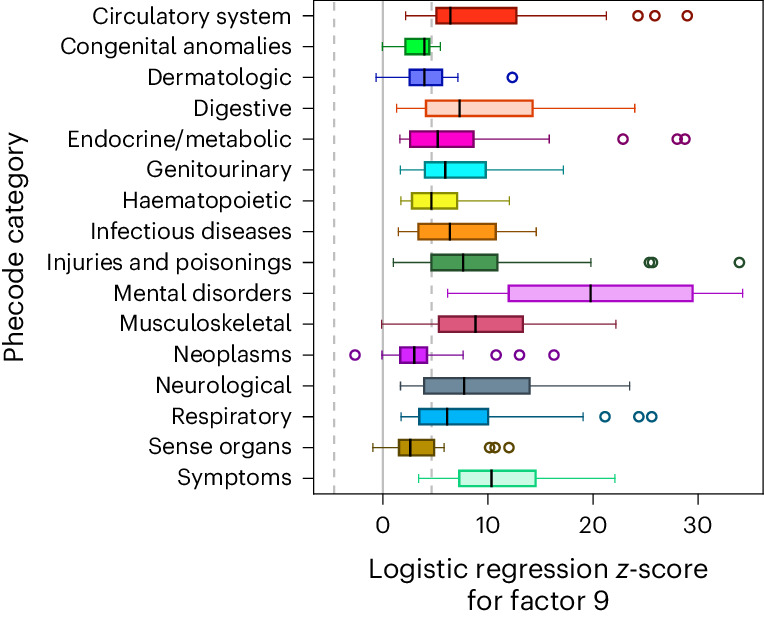
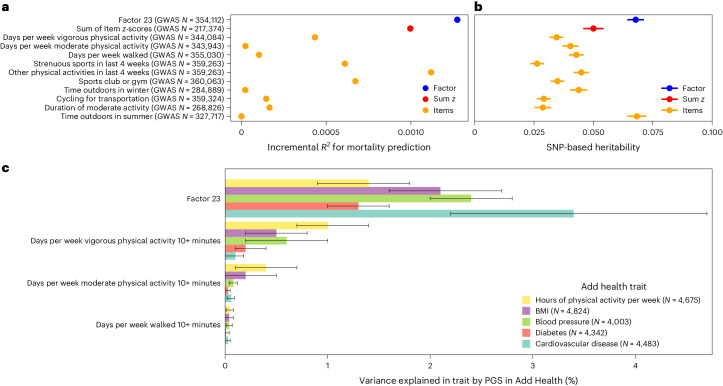
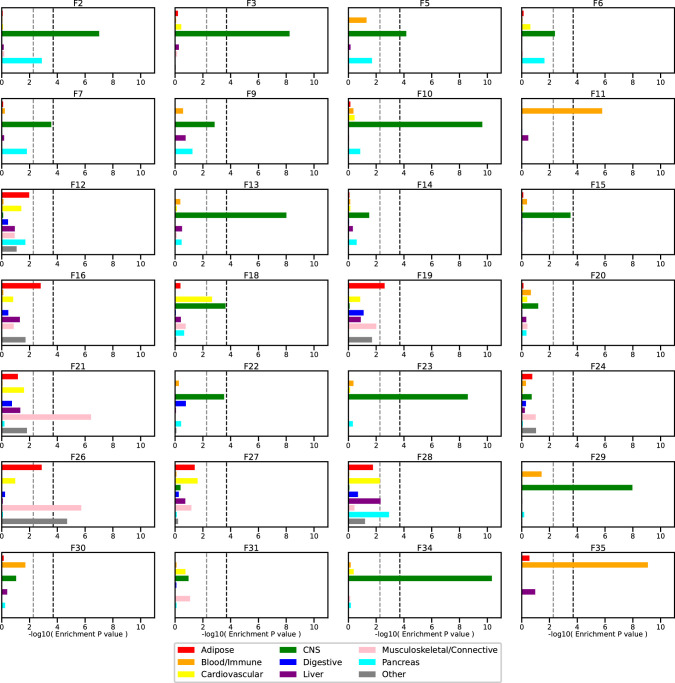
Data within biobanks capture broad yet detailed indices of human variation, but biobank-wide insights can be difficult to extract due to complexity and scale. Here, using large-scale factor analysis, we distill hundreds of variables (diagnoses, assessments and survey items) into 35 latent constructs, using data from unrelated individuals with predominantly estimated European genetic ancestry in UK Biobank. These factors recapitulate known disease classifications, disentangle elements of socioeconomic status, highlight the relevance of psychiatric constructs to health and improve measurement of pro-health behaviours. We go on to demonstrate the power of this approach to clarify genetic signal, enhance discovery and identify associations between underlying phenotypic structure and health outcomes. In building a deeper understanding of ways in which constructs such as socioeconomic status, trauma, or physical activity are structured in the dataset, we emphasize the importance of considering the interwoven nature of the human phenome when evaluating public health patterns.
© 2024. The Author(s).
Conflict of interest statement
C.E.C. is currently an employee of Novartis. R.W. is a research fellow at AnalytiXIN, which is a consortium of health-data organizations, industry partners and university partners in Indiana primarily funded through the Lilly Endowment, IU Health and Eli Lilly and Company. B.M.N. is a member of the scientific advisory board at Deep Genomics and Neumora, and a consultant of the scientific advisory board of Camp4 Therapeutics. R.K.W. has received honoraria from the Jackson Laboratory and sponsored travel from the Russell Sage Foundation in the past 36 months. G.D.S. reports Scientific Advisory Board Membership for Relation Therapeutics and Insitro. The remaining authors declare no competing interests.
Figures









References
MeSH terms
Grants and funding
- R01 MH124851/MH/NIMH NIH HHS/United States
- R37 MH107649/MH/NIMH NIH HHS/United States
- R01 HD060726/HD/NICHD NIH HHS/United States
- R01 HD073342/HD/NICHD NIH HHS/United States
- 5R37MH107649/U.S. Department of Health & Human Services | NIH | National Institute of Mental Health (NIMH)
- R01MH101244/U.S. Department of Health & Human Services | NIH | National Institute of Mental Health (NIMH)
- R01 MH101244/MH/NIMH NIH HHS/United States
- R01MH124851/U.S. Department of Health & Human Services | NIH | National Institute of Mental Health (NIMH)
- P01 HD031921/HD/NICHD NIH HHS/United States
- NNF21SA0072102/Novo Nordisk Fonden (Novo Nordisk Foundation)
LinkOut - more resources
Full Text Sources
