

Polyamine and EIF5A hypusination downstream of c-Myc confers targeted therapy resistance in BRAF mutant melanoma
- PMID: 38965534
- PMCID: PMC11223307
- DOI: 10.1186/s12943-024-02031-w
Polyamine and EIF5A hypusination downstream of c-Myc confers targeted therapy resistance in BRAF mutant melanoma
Abstract
Background: BRAF inhibitors are widely employed in the treatment of melanoma with the BRAF V600E mutation. However, the development of resistance compromises their therapeutic efficacy. Diverse genomic and transcriptomic alterations are found in BRAF inhibitor resistant melanoma, posing a pressing need for convergent, druggable target that reverse therapy resistant tumor with different resistance mechanisms.
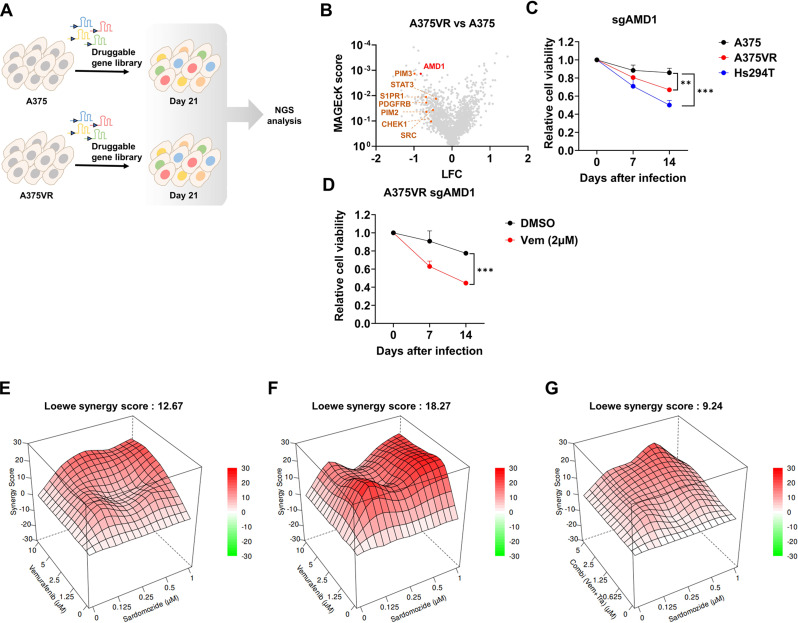
Methods: CRISPR-Cas9 screens were performed to identify novel target gene whose inhibition selectively targets A375VR, a BRAF V600E mutant cell line with acquired resistance to vemurafenib. Various in vitro and in vivo assays, including cell competition assay, water soluble tetrazolium (WST) assay, live-dead assay and xenograft assay were performed to confirm synergistic cell death. Liquid Chromatography-Mass Spectrometry analyses quantified polyamine biosynthesis and changes in proteome in vemurafenib resistant melanoma. EIF5A hypusination dependent protein translation and subsequent changes in mitochondrial biogenesis and activity were assayed by O-propargyl-puromycin labeling assay, mitotracker, mitoSOX labeling and seahorse assay. Bioinformatics analyses were used to identify the association of polyamine biosynthesis with BRAF inhibitor resistance and poor prognosis in melanoma patient cohorts.
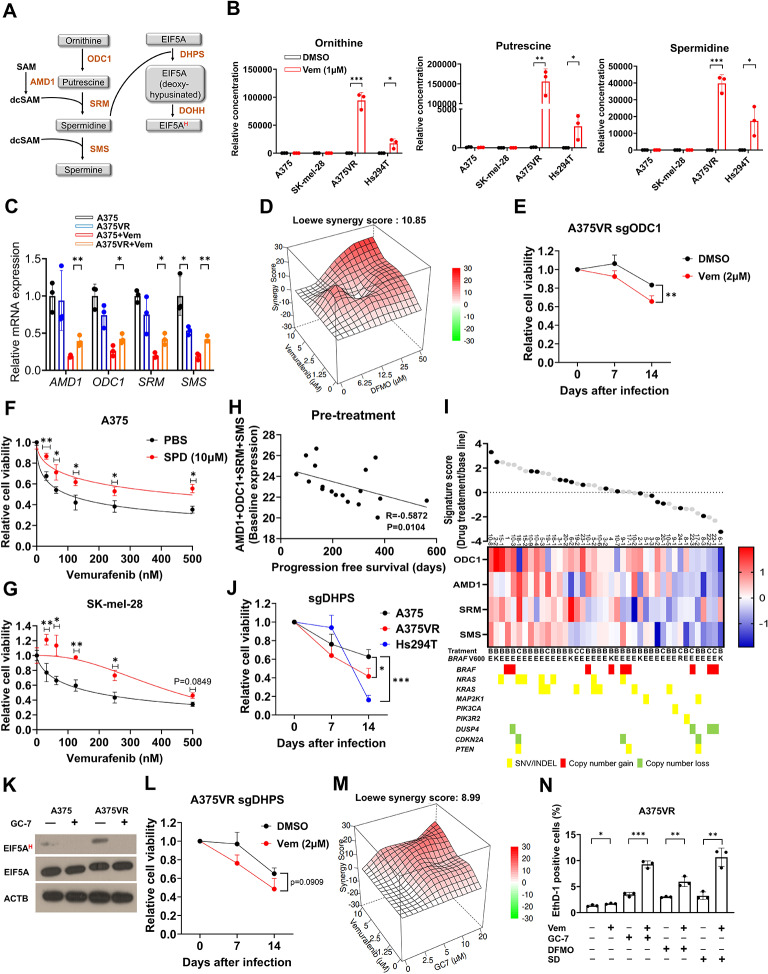
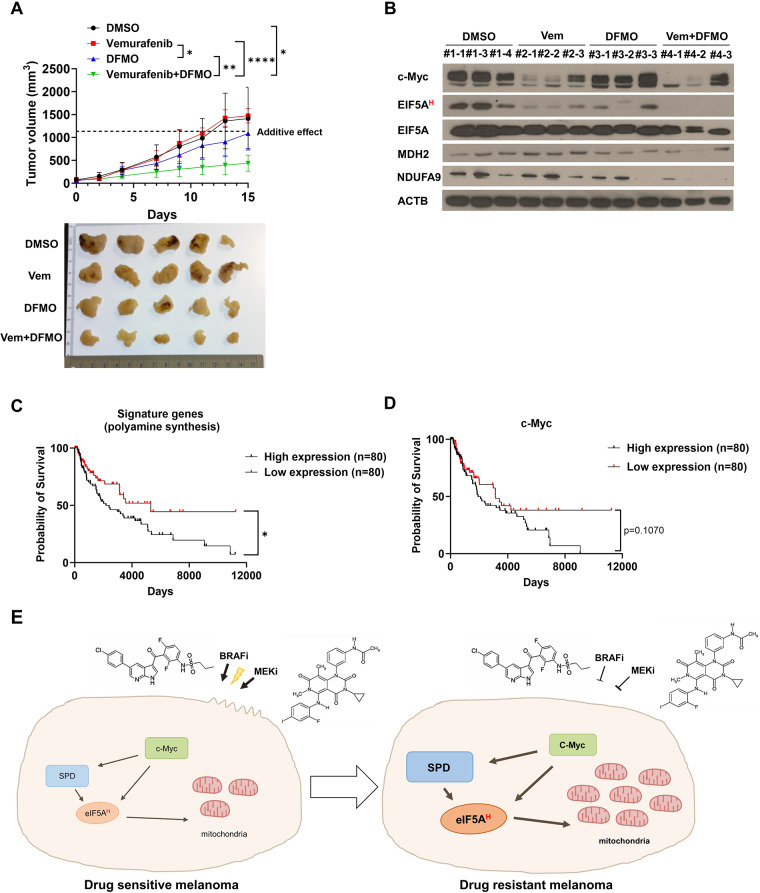
Results: We elucidate the role of polyamine biosynthesis and its regulatory mechanisms in promoting BRAF inhibitor resistance. Leveraging CRISPR-Cas9 screens, we identify AMD1 (S-adenosylmethionine decarboxylase 1), a critical enzyme for polyamine biosynthesis, as a druggable target whose inhibition reduces vemurafenib resistance. Metabolomic and proteomic analyses reveal that polyamine biosynthesis is upregulated in vemurafenib-resistant cancer, resulting in enhanced EIF5A hypusination, translation of mitochondrial proteins and oxidative phosphorylation. We also identify that sustained c-Myc levels in vemurafenib-resistant cancer are responsible for elevated polyamine biosynthesis. Inhibition of polyamine biosynthesis or c-Myc reversed vemurafenib resistance both in vitro cell line models and in vivo in a xenograft model. Polyamine biosynthesis signature is associated with poor prognosis and shorter progression free survival after BRAF/MAPK inhibitor treatment in melanoma cohorts, highlighting the clinical relevance of our findings.
Conclusions: Our findings delineate the molecular mechanisms involving polyamine-EIF5A hypusination-mitochondrial respiration pathway conferring BRAF inhibitor resistance in melanoma. These targets will serve as effective therapeutic targets that can maximize the therapeutic efficacy of existing BRAF inhibitors.
Keywords: BRAF; Drug resistance; Hypusination; Melanoma; Mitochondria; Polyamine; Vemurafenib; c-Myc.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, Mina L, Dragovich T, Gimbel M, Mahmoud F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018;11:7095–107. doi: 10.2147/OTT.S182721. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
