

Rapid identification and subsequent contextualization of an outbreak of methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit using nanopore sequencing
- PMID: 38967541
- PMCID: PMC11316549
- DOI: 10.1099/mgen.0.001273
Rapid identification and subsequent contextualization of an outbreak of methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit using nanopore sequencing
Abstract
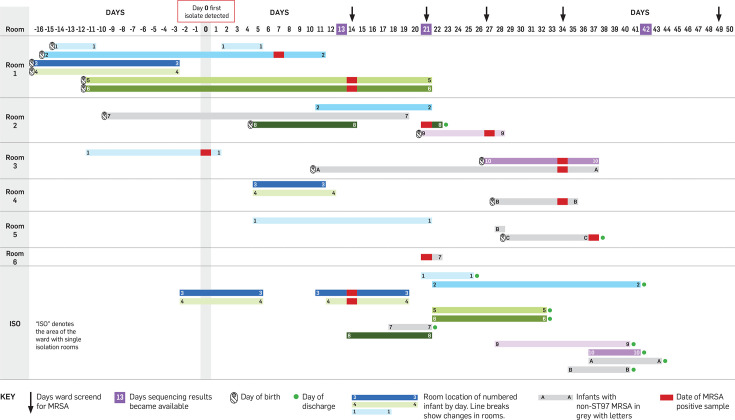
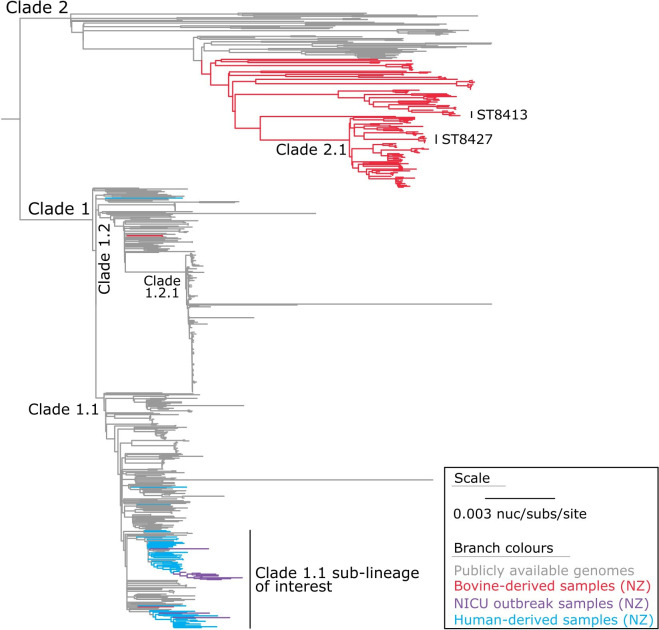
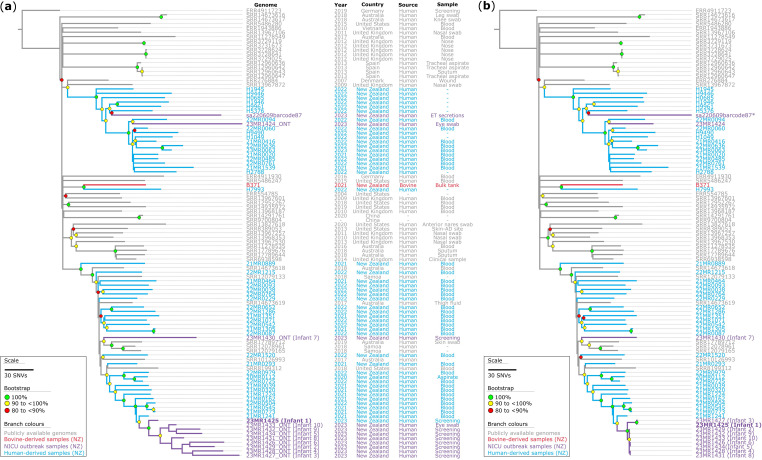
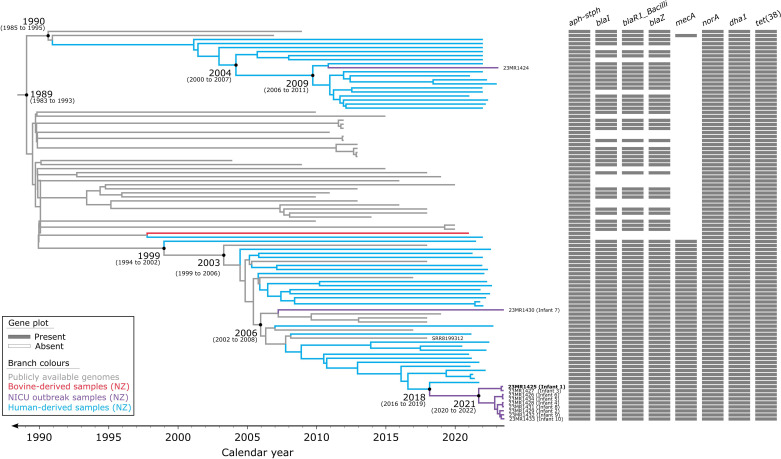
Outbreaks of methicillin-resistant Staphylococcus aureus (MRSA) are well described in the neonatal intensive care unit (NICU) setting. Genomics has revolutionized the investigation of such outbreaks; however, to date, this has largely been completed retrospectively and has typically relied on short-read platforms. In 2022, our laboratory established a prospective genomic surveillance system using Oxford Nanopore Technologies sequencing for rapid outbreak detection. Herein, using this system, we describe the detection and control of an outbreak of sequence-type (ST)97 MRSA in our NICU. The outbreak was identified 13 days after the first MRSA-positive culture and at a point where there were only two known cases. Ward screening rapidly defined the extent of the outbreak, with six other infants found to be colonized. There was minimal transmission once the outbreak had been detected and appropriate infection control measures had been instituted; only two further ST97 cases were detected, along with three unrelated non-ST97 MRSA cases. To contextualize the outbreak, core-genome single-nucleotide variants were identified for phylogenetic analysis after de novo assembly of nanopore data. Comparisons with global (n=45) and national surveillance (n=35) ST97 genomes revealed the stepwise evolution of methicillin resistance within this ST97 subset. A distinct cluster comprising nine of the ten ST97-IVa genomes from the NICU was identified, with strains from 2020 to 2022 national surveillance serving as outgroups to this cluster. One ST97-IVa genome presumed to be part of the outbreak formed an outgroup and was retrospectively excluded. A second phylogeny was created using Illumina sequencing, which considerably reduced the branch lengths of the NICU isolates on the phylogenetic tree. However, the overall tree topology and conclusions were unchanged, with the exception of the NICU outbreak cluster, where differences in branch lengths were observed. This analysis demonstrated the ability of a nanopore-only prospective genomic surveillance system to rapidly identify and contextualize an outbreak of MRSA in a NICU.
Keywords: antibiotic resistance; genomic surveillance; infection control; outbreak detection; phylogenetic analysis.
Conflict of interest statement
The authors declare that there are no conflicts of financial, general or institutional competing interests.
Figures
Similar articles
-
Whole-genome sequencing data-based modeling for the investigation of an outbreak of community-associated methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit in Hong Kong.Eur J Clin Microbiol Infect Dis. 2019 Mar;38(3):563-573. doi: 10.1007/s10096-018-03458-y. Epub 2019 Jan 24. Eur J Clin Microbiol Infect Dis. 2019. PMID: 30680562
-
A Complete Genome Screening Program of Clinical Methicillin-Resistant Staphylococcus aureus Isolates Identifies the Origin and Progression of a Neonatal Intensive Care Unit Outbreak.J Clin Microbiol. 2019 Nov 22;57(12):e01261-19. doi: 10.1128/JCM.01261-19. Print 2019 Dec. J Clin Microbiol. 2019. PMID: 31578260 Free PMC article.
-
Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak.N Engl J Med. 2012 Jun 14;366(24):2267-75. doi: 10.1056/NEJMoa1109910. N Engl J Med. 2012. PMID: 22693998 Free PMC article.
-
Methicillin-resistant Staphylococcus aureus (MRSA) clusters in neonatal intensive care units (NICUs) and other neonatal units in New York State (NYS), 2001 to 2017.Am J Infect Control. 2024 Apr;52(4):424-435. doi: 10.1016/j.ajic.2023.09.015. Epub 2023 Sep 29. Am J Infect Control. 2024. PMID: 37778709 Review.
-
A model to implement genomic medicine in the neonatal intensive care unit.J Perinatol. 2023 Feb;43(2):248-252. doi: 10.1038/s41372-022-01428-z. Epub 2022 Jun 24. J Perinatol. 2023. PMID: 35750755 Free PMC article. Review.
Cited by
-
The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing.Antimicrob Resist Infect Control. 2025 Feb 7;14(1):6. doi: 10.1186/s13756-025-01529-2. Antimicrob Resist Infect Control. 2025. PMID: 39920743 Free PMC article.
-
The Host Adaptation of Staphylococcus aureus to Farmed Ruminants in New Zealand, With Special Reference to Clonal Complex 1.Environ Microbiol Rep. 2025 Jun;17(3):e70087. doi: 10.1111/1758-2229.70087. Environ Microbiol Rep. 2025. PMID: 40329524 Free PMC article.
References
-
- Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, et al. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997-1999. Clin Infect Dis. 2001;32:S114–S132. doi: 10.1086/320184. - DOI - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical