

Elevating PLK1 overcomes BETi resistance in prostate cancer via triggering BRD4 phosphorylation-dependent degradation in mitosis
- PMID: 38968071
- PMCID: PMC11334074
- DOI: 10.1016/j.celrep.2024.114431
Elevating PLK1 overcomes BETi resistance in prostate cancer via triggering BRD4 phosphorylation-dependent degradation in mitosis
Abstract
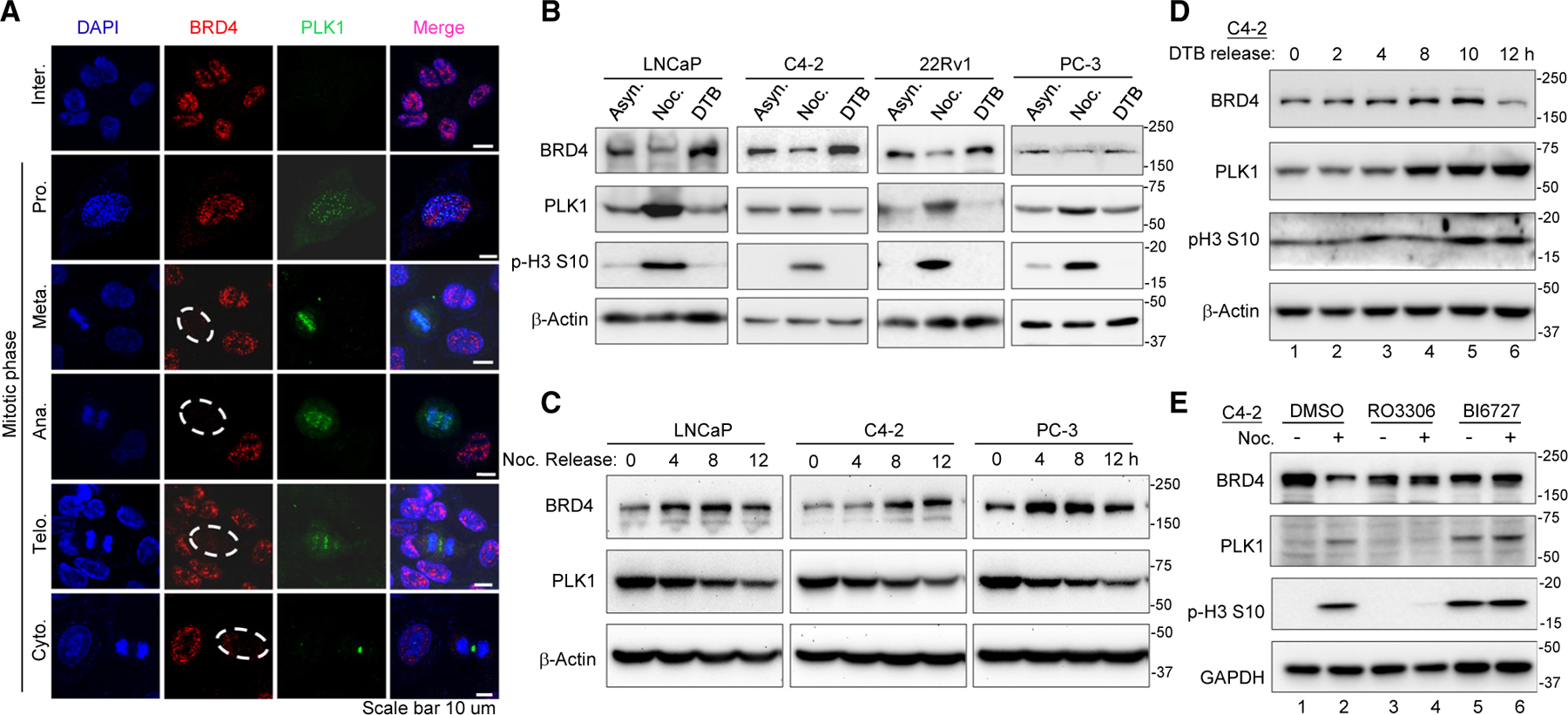
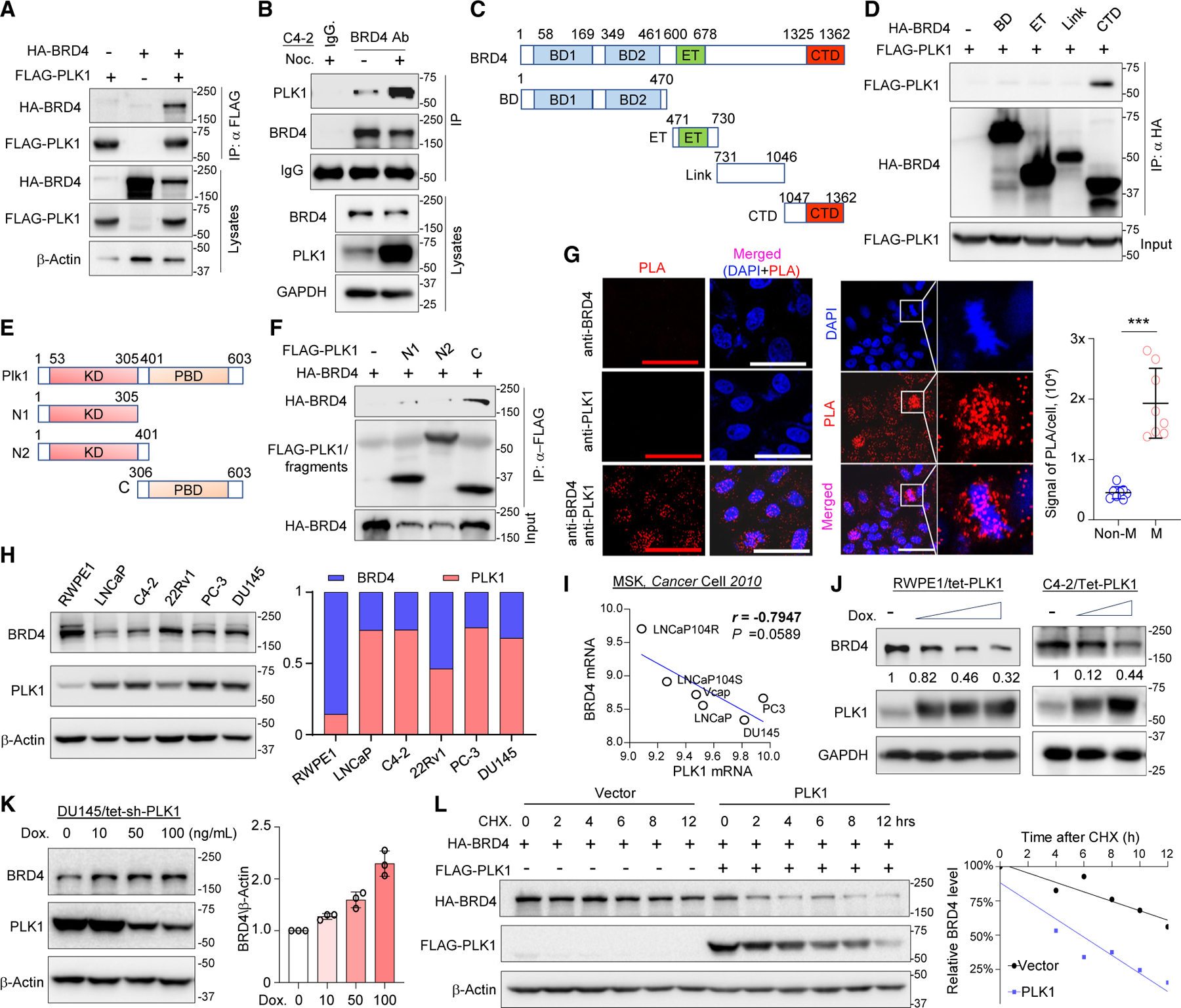
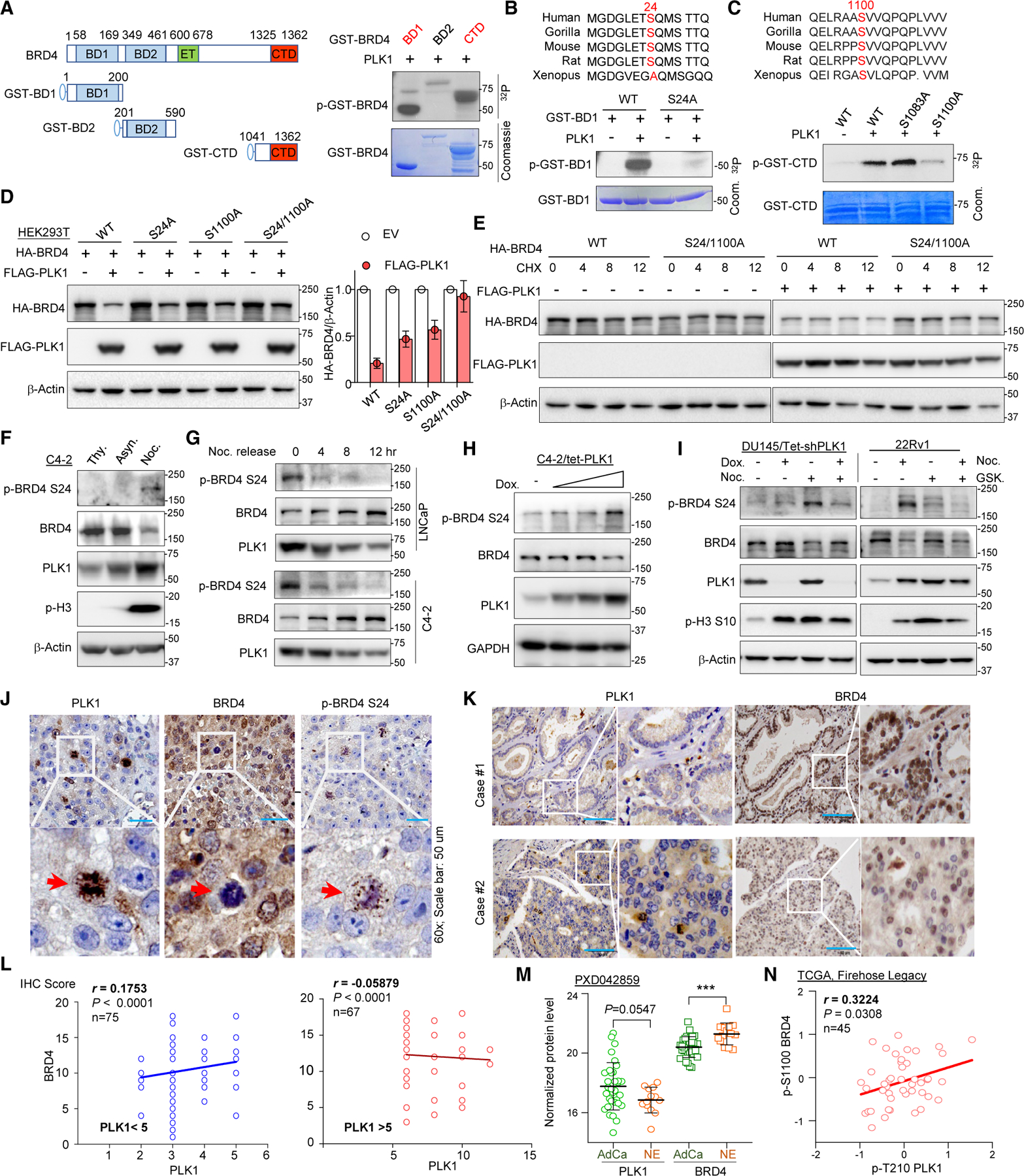
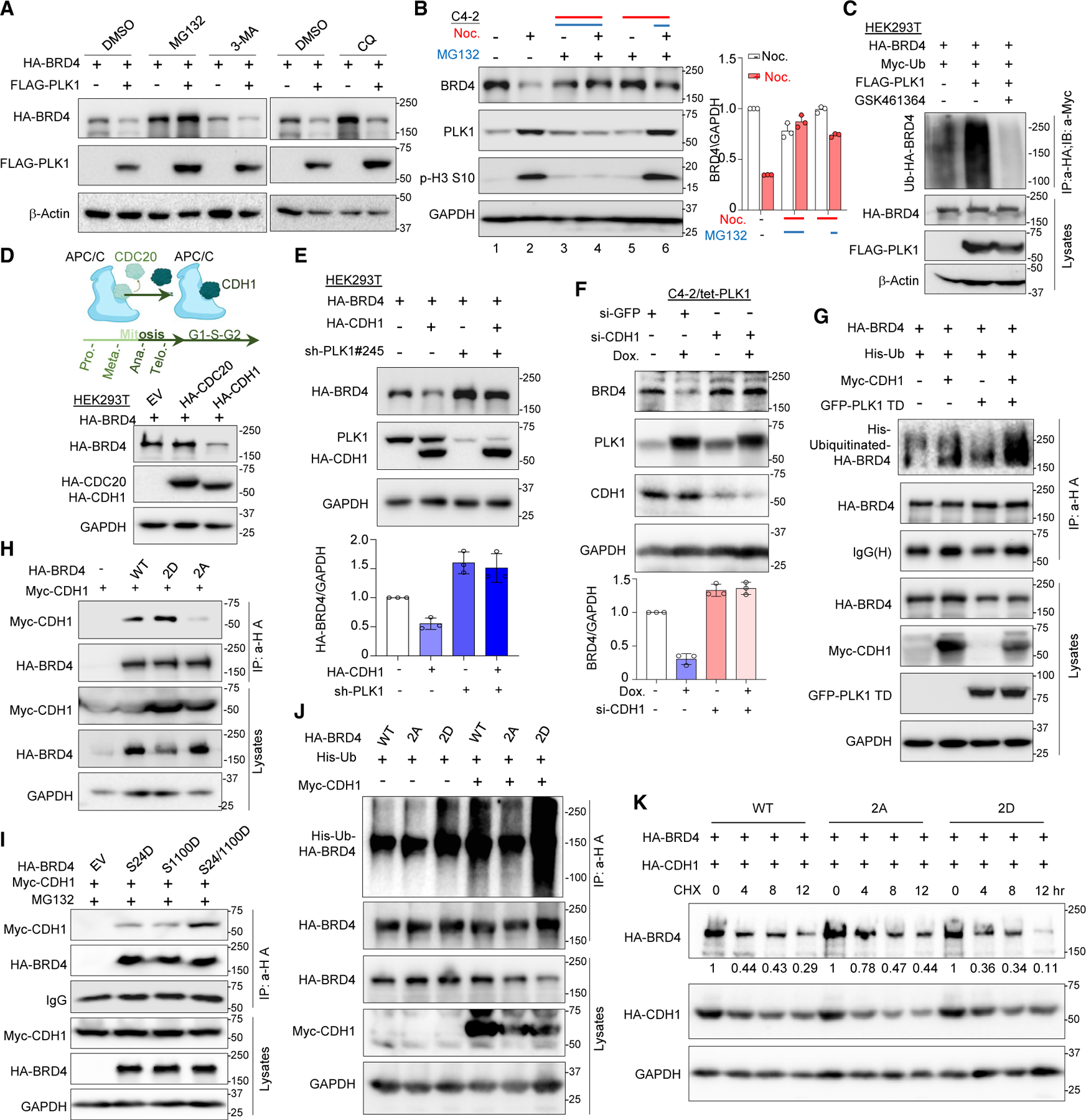
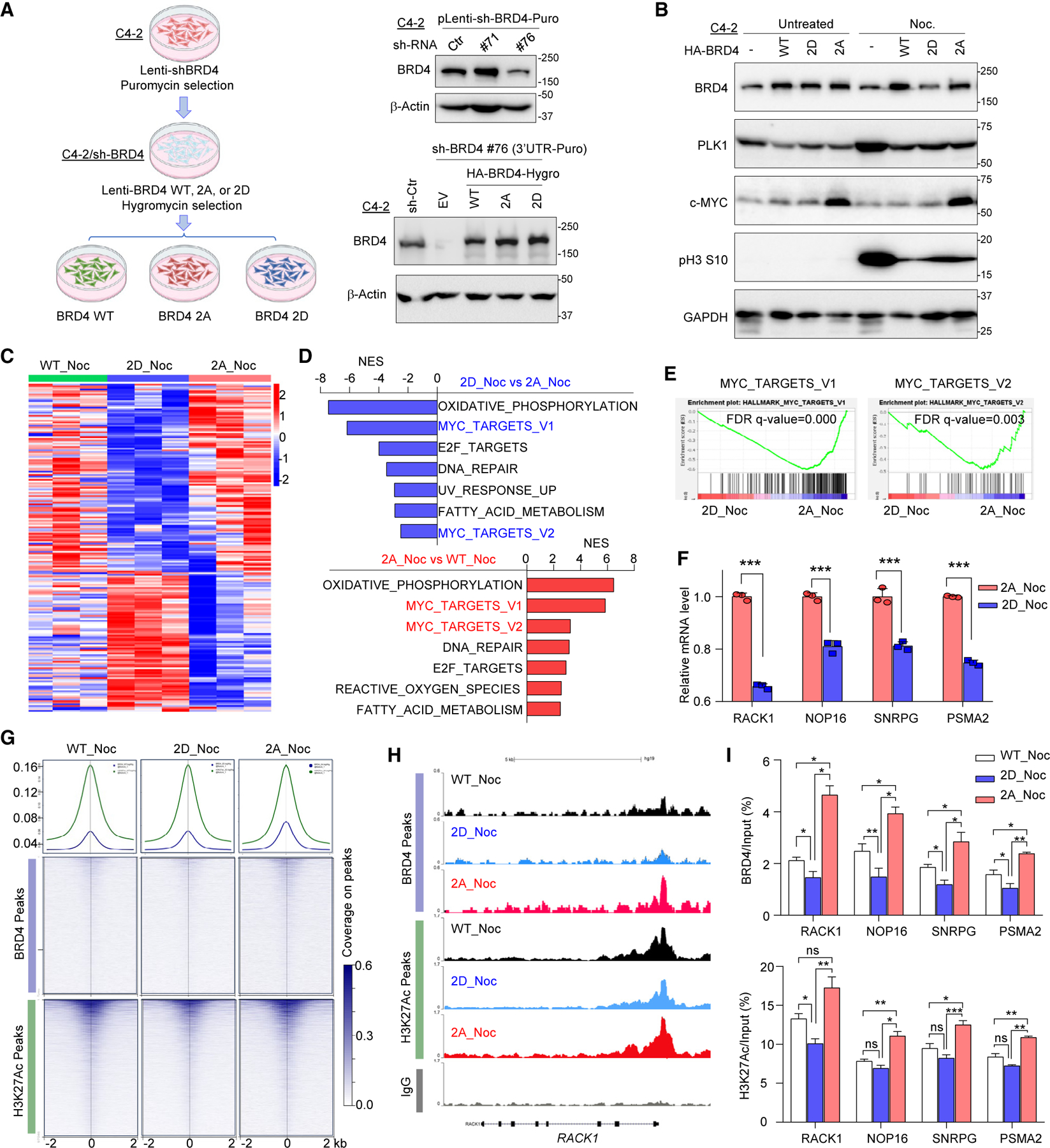
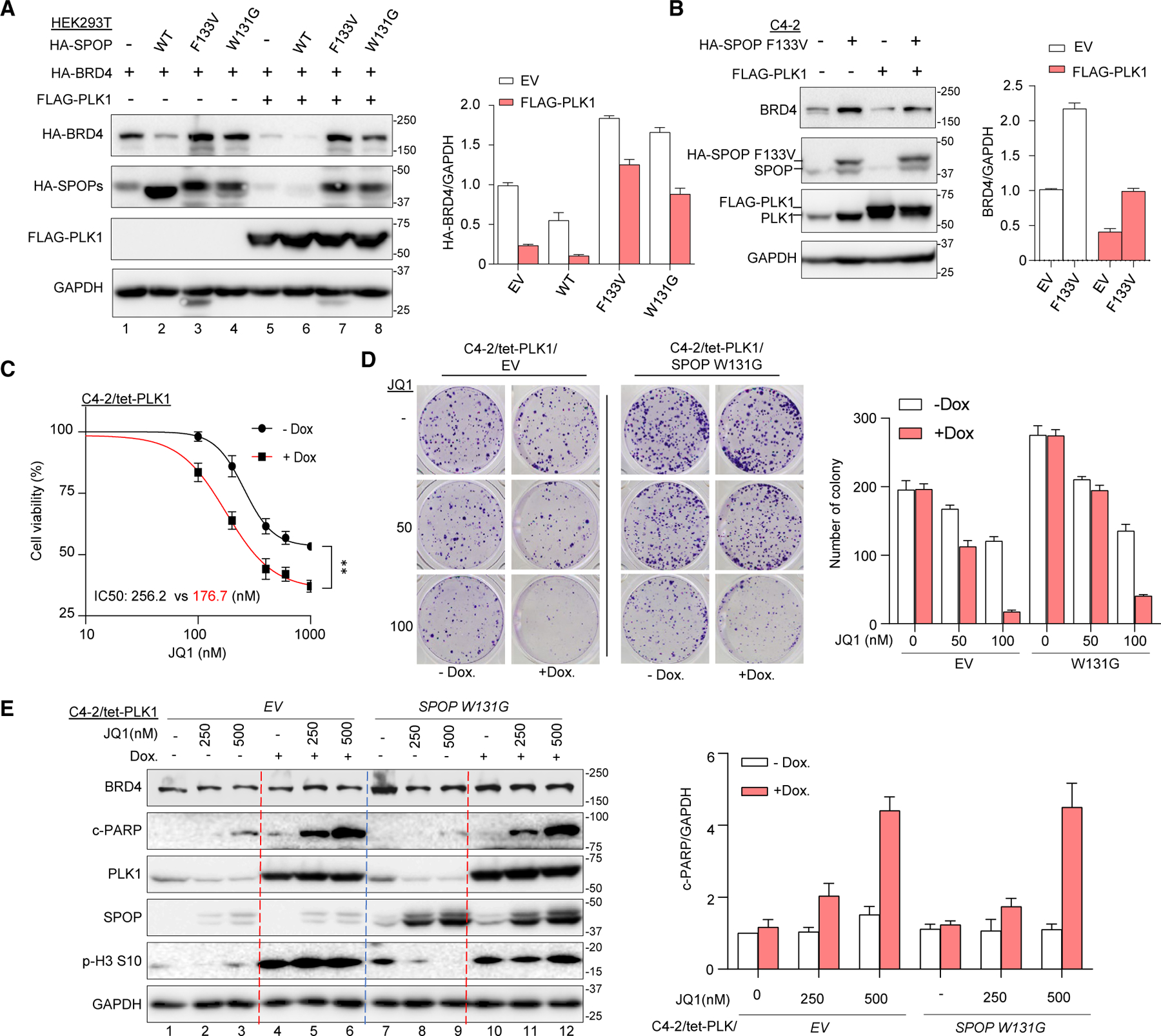
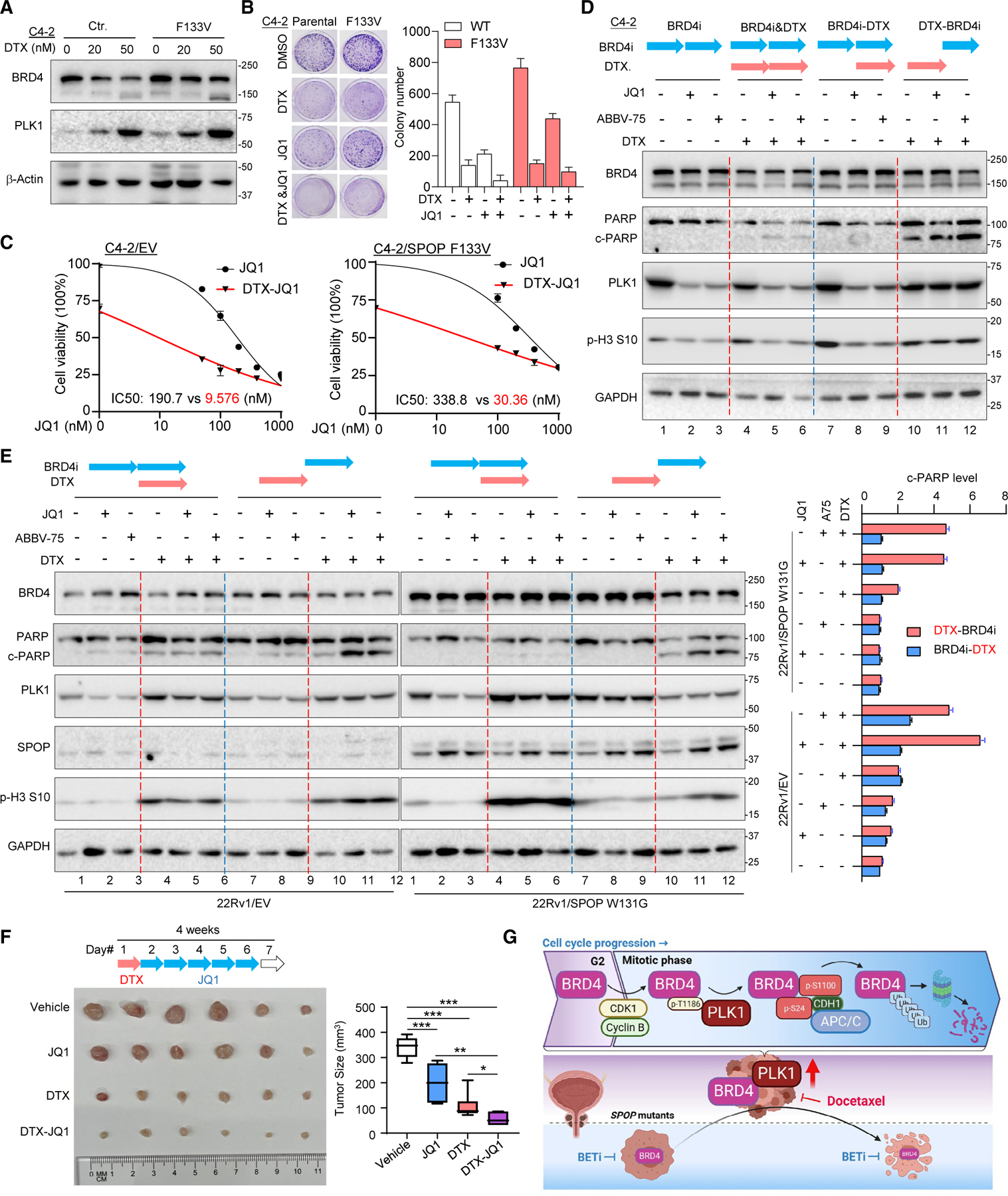
Bromodomain-containing protein 4 (BRD4) has emerged as a promising therapeutic target in prostate cancer (PCa). Understanding the mechanisms of BRD4 stability could enhance the clinical response to BRD4-targeted therapy. In this study, we report that BRD4 protein levels are significantly decreased during mitosis in a PLK1-dependent manner. Mechanistically, we show that BRD4 is primarily phosphorylated at T1186 by the CDK1/cyclin B complex, recruiting PLK1 to phosphorylate BRD4 at S24/S1100, which are recognized by the APC/CCdh1 complex for proteasome pathway degradation. We find that PLK1 overexpression lowers SPOP mutation-stabilized BRD4, consequently rendering PCa cells re-sensitized to BRD4 inhibitors. Intriguingly, we report that sequential treatment of docetaxel and JQ1 resulted in significant inhibition of PCa. Collectively, the results support that PLK1-phosphorylated BRD4 triggers its degradation at M phase. Sequential treatment of docetaxel and JQ1 overcomes BRD4 accumulation-associated bromodomain and extra-terminal inhibitor (BETi) resistance, which may shed light on the development of strategies to treat PCa.
Keywords: APC/C(Cdh1); BET inhibitor resistance; BRD4; CP: Cancer; CP: Molecular biology; PLK1; phosphorylation; prostate cancer.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
