

Estimating error rates for single molecule protein sequencing experiments
- PMID: 38968291
- PMCID: PMC11253918
- DOI: 10.1371/journal.pcbi.1012258
Estimating error rates for single molecule protein sequencing experiments
Abstract
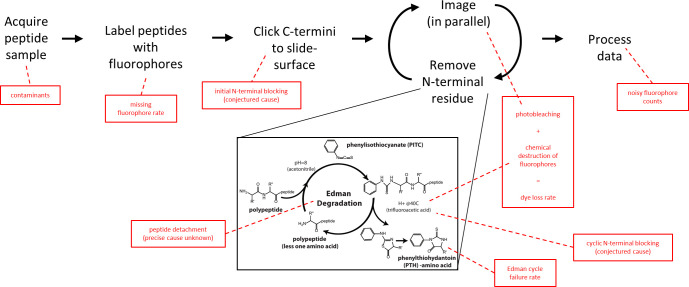
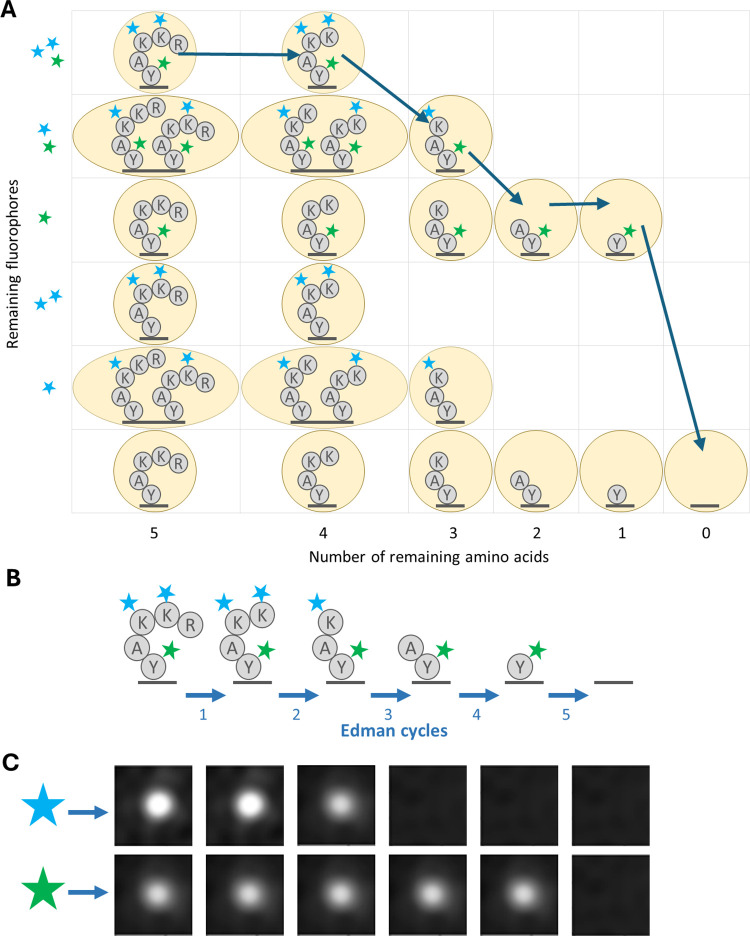
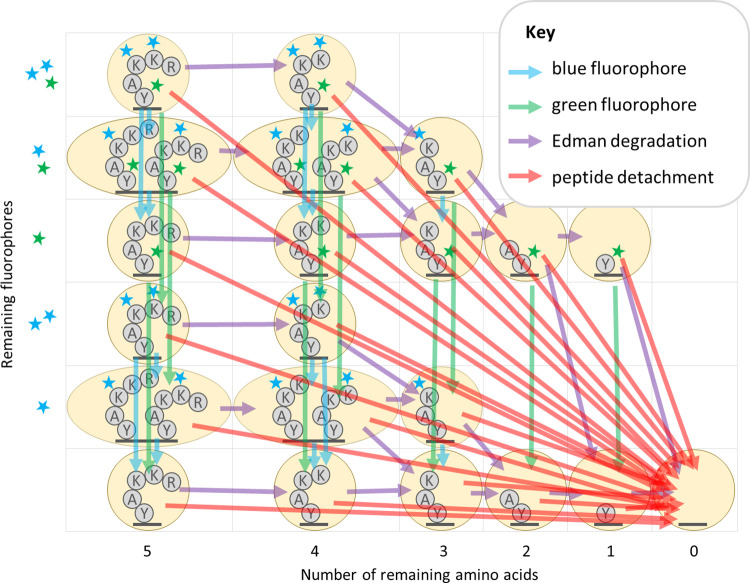
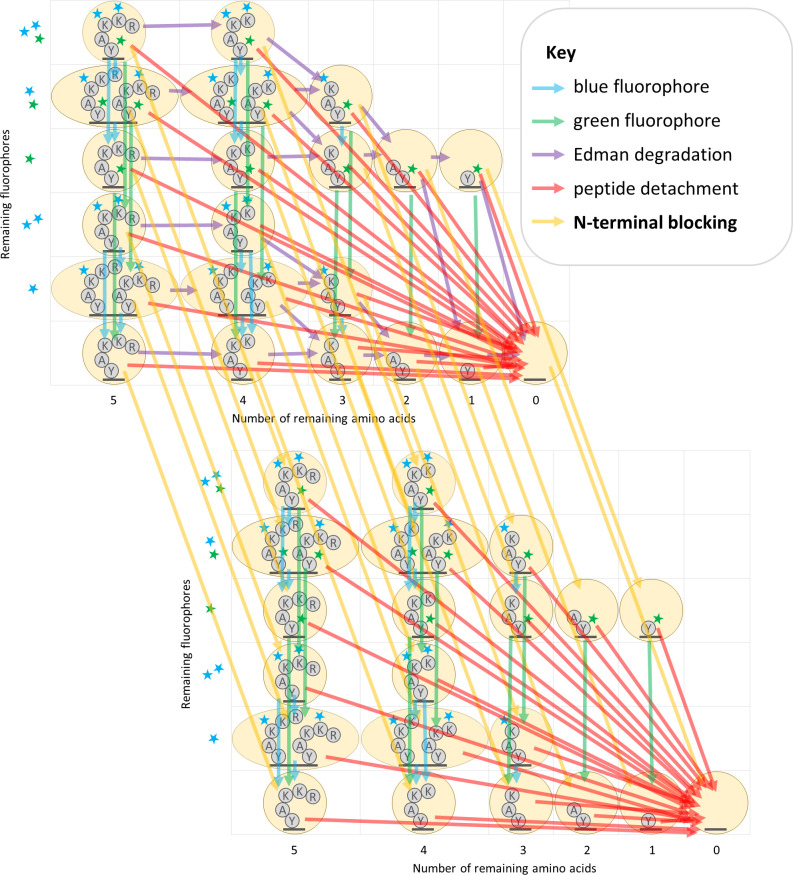
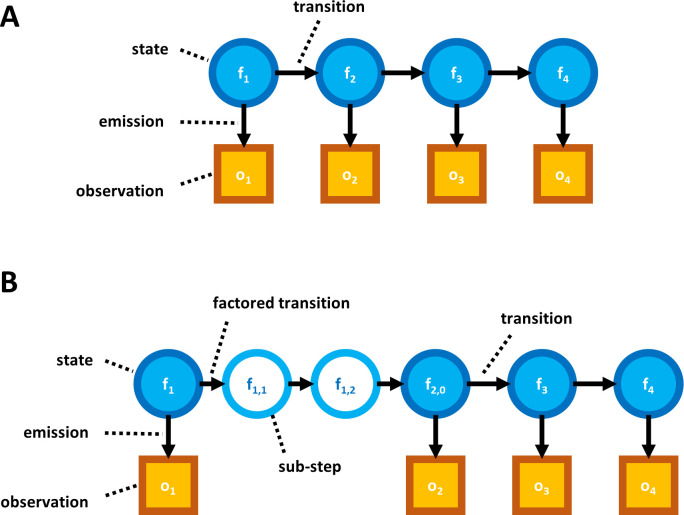
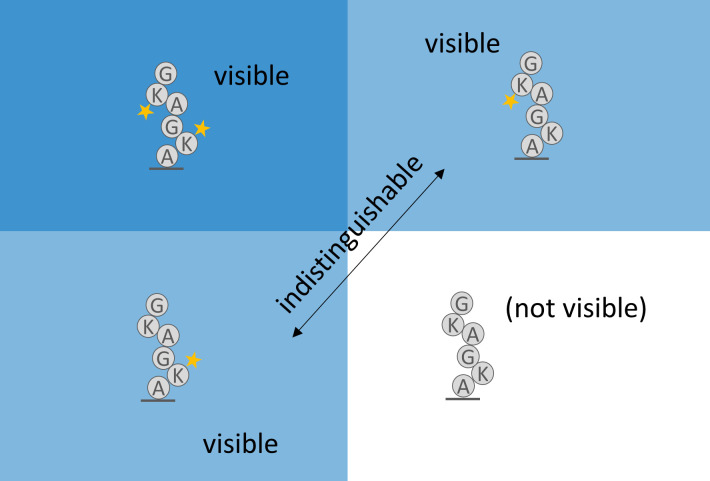
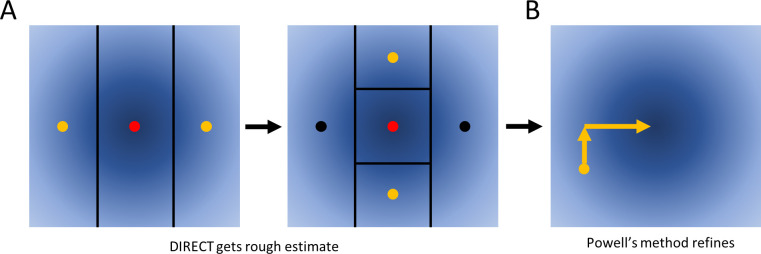
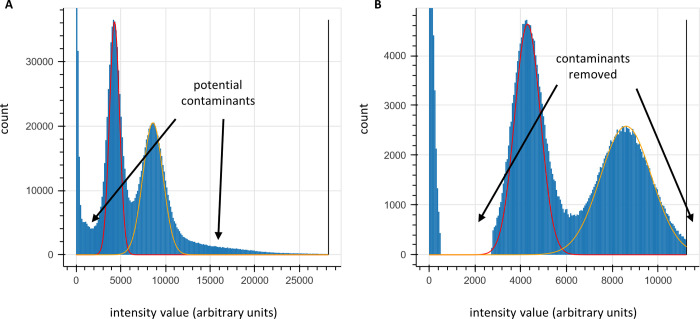
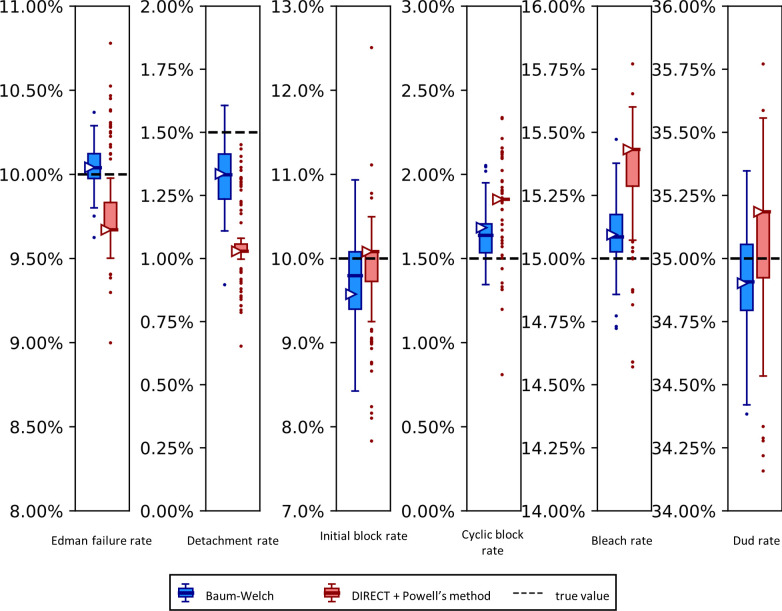
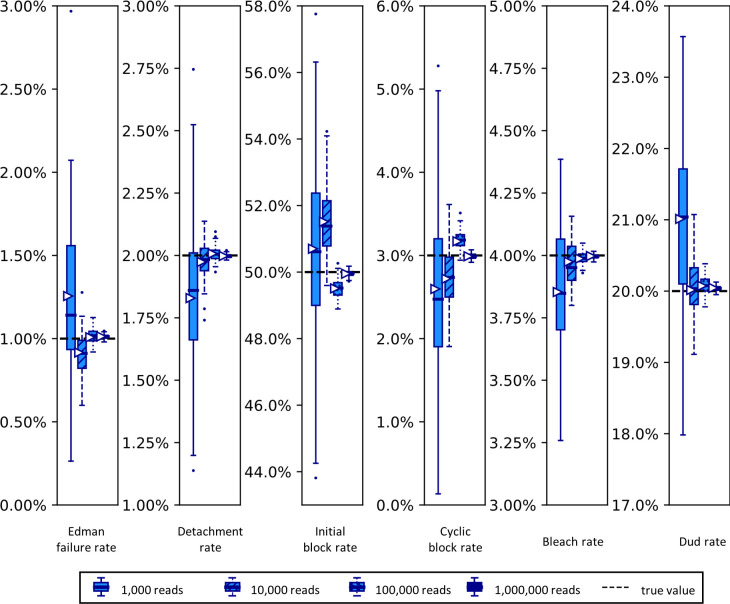
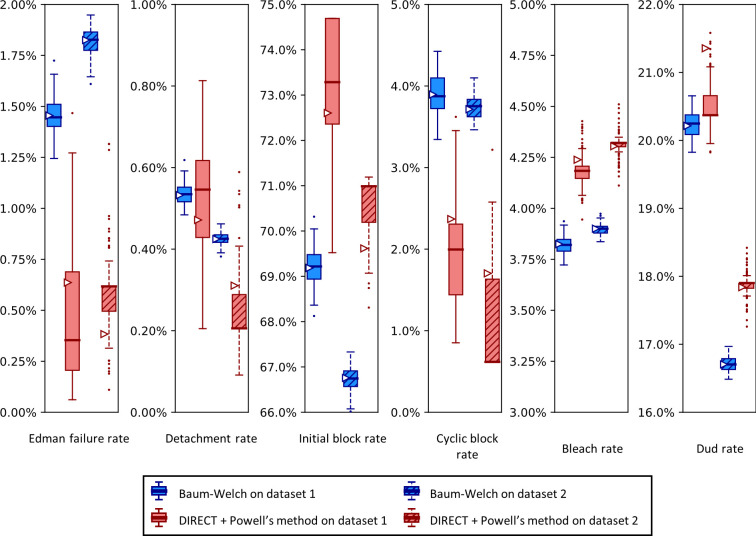
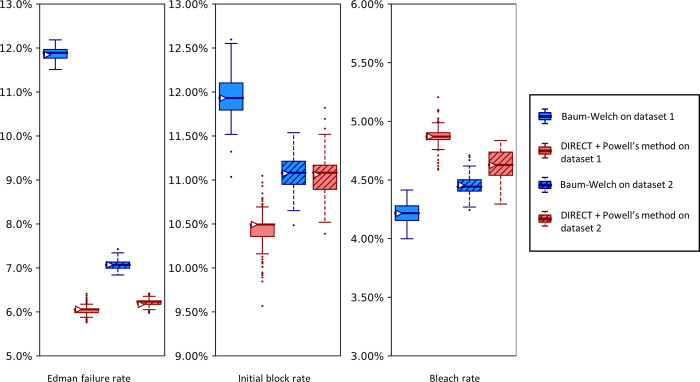
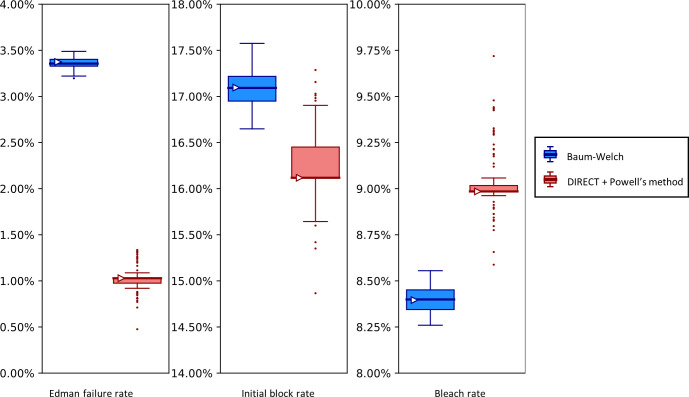
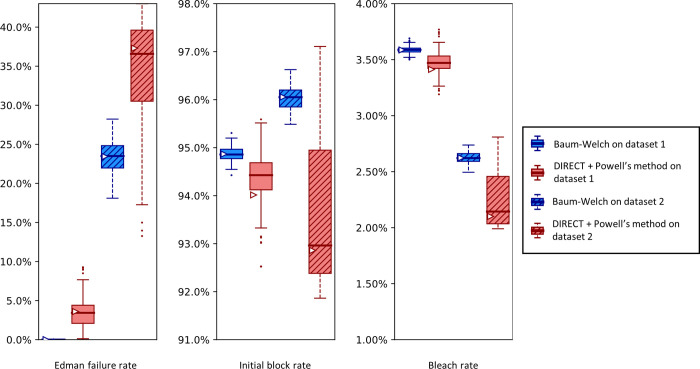
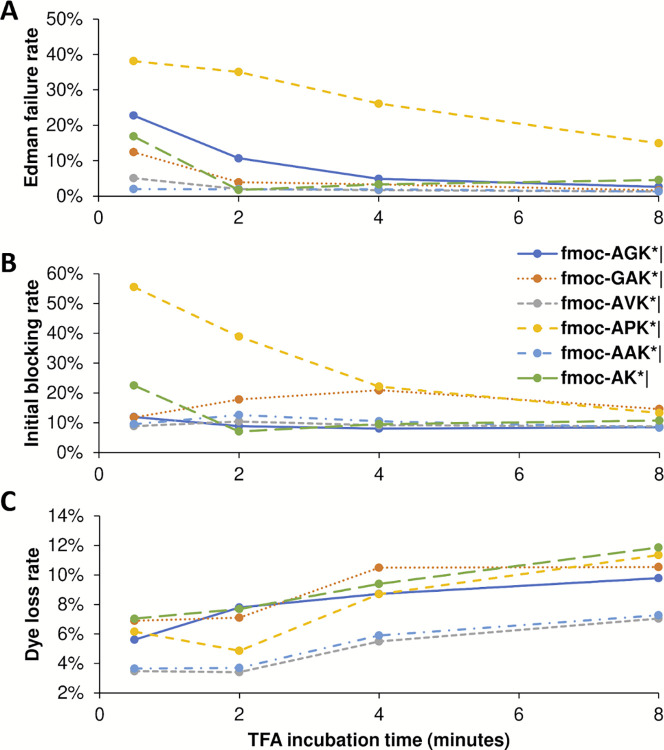
The practical application of new single molecule protein sequencing (SMPS) technologies requires accurate estimates of their associated sequencing error rates. Here, we describe the development and application of two distinct parameter estimation methods for analyzing SMPS reads produced by fluorosequencing. A Hidden Markov Model (HMM) based approach, extends whatprot, where we previously used HMMs for SMPS peptide-read matching. This extension offers a principled approach for estimating key parameters for fluorosequencing experiments, including missed amino acid cleavages, dye loss, and peptide detachment. Specifically, we adapted the Baum-Welch algorithm, a standard technique to estimate transition probabilities for an HMM using expectation maximization, but modified here to estimate a small number of parameter values directly rather than estimating every transition probability independently. We demonstrate a high degree of accuracy on simulated data, but on experimental datasets, we observed that the model needed to be augmented with an additional error type, N-terminal blocking. This, in combination with data pre-processing, results in reasonable parameterizations of experimental datasets that agree with controlled experimental perturbations. A second independent implementation using a hybrid of DIRECT and Powell's method to reduce the root mean squared error (RMSE) between simulations and the real dataset was also developed. We compare these methods on both simulated and real data, finding that our Baum-Welch based approach outperforms DIRECT and Powell's method by most, but not all, criteria. Although some discrepancies between the results exist, we also find that both approaches provide similar error rate estimates from experimental single molecule fluorosequencing datasets.
Copyright: © 2024 Smith et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: A.M.B. and E.M.M. are co-founders and shareholders of Erisyon, Inc., and are co-inventors on granted patents or pending patent applications related to single-molecule protein sequencing. A.M.B. serves on the board of directors and E.M.M. serves on the scientific advisory board. M.B.S., K.V., T.B., H.D.S., J.H.M., T.M.F., C.M., and A.M.B. are affiliated with Erisyon, Inc., as employees or shareholders. H.D.S. is currently employed by UT Austin with funding from a Sponsored Research Agreement from Erisyon, Inc.
Figures
Update of
-
Estimating error rates for single molecule protein sequencing experiments.bioRxiv [Preprint]. 2023 Jul 19:2023.07.18.549591. doi: 10.1101/2023.07.18.549591. bioRxiv. 2023. Update in: PLoS Comput Biol. 2024 Jul 5;20(7):e1012258. doi: 10.1371/journal.pcbi.1012258. PMID: 37502879 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
