

Saturation genome editing of BAP1 functionally classifies somatic and germline variants
- PMID: 38969833
- PMCID: PMC11250367
- DOI: 10.1038/s41588-024-01799-3
Saturation genome editing of BAP1 functionally classifies somatic and germline variants
Abstract
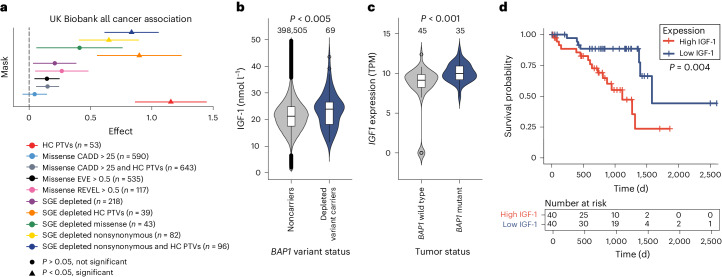
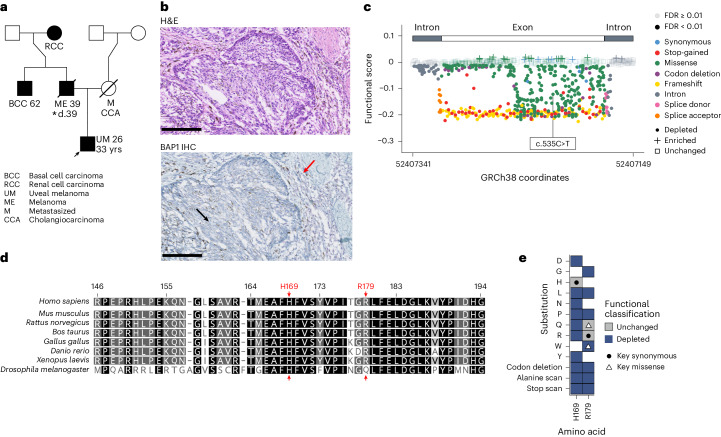
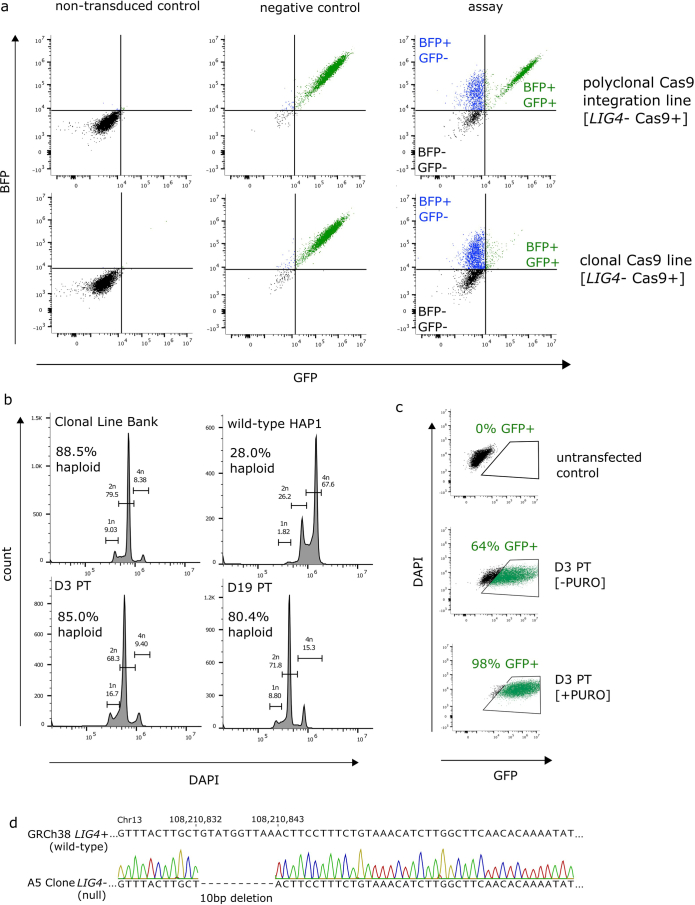
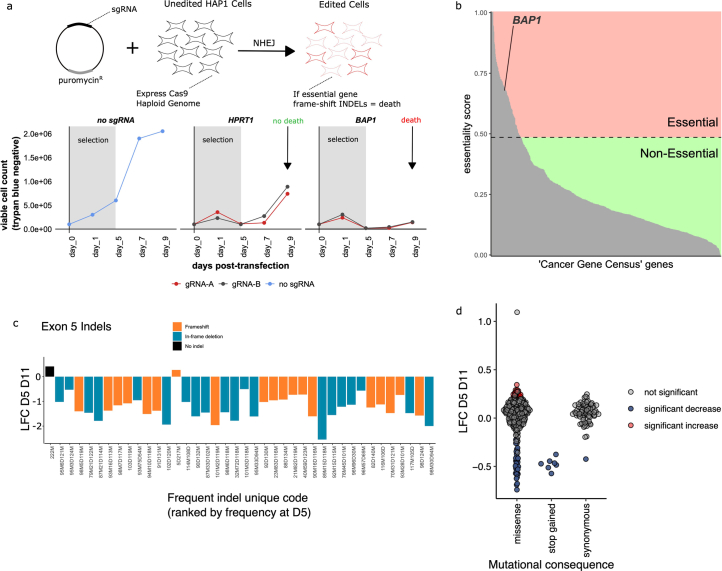
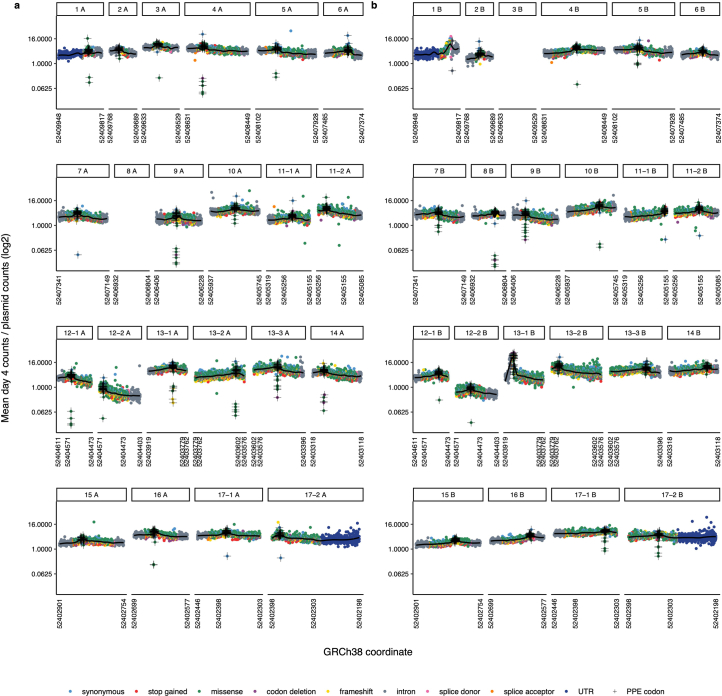
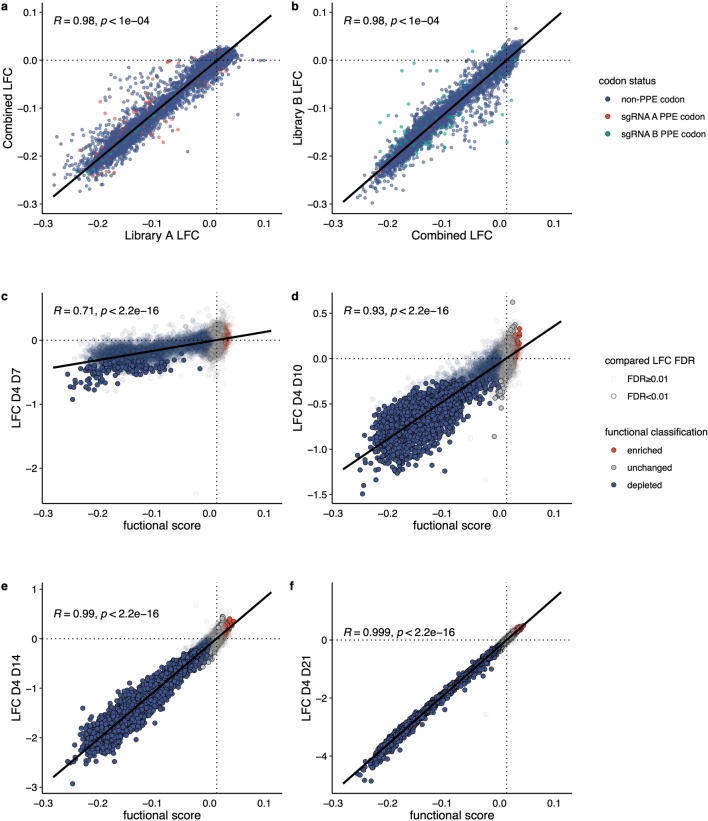
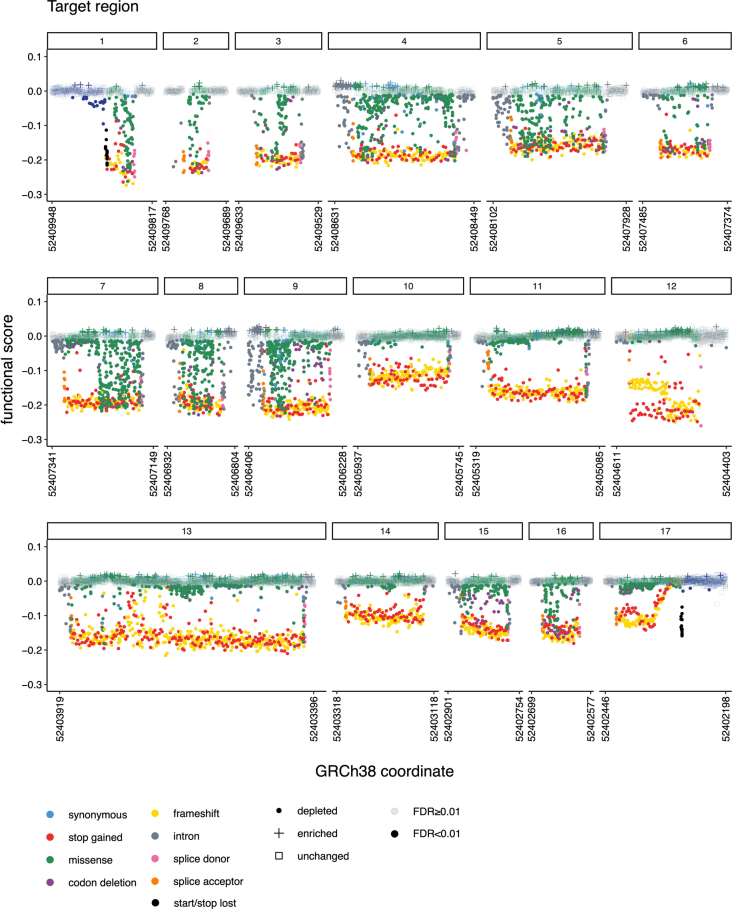
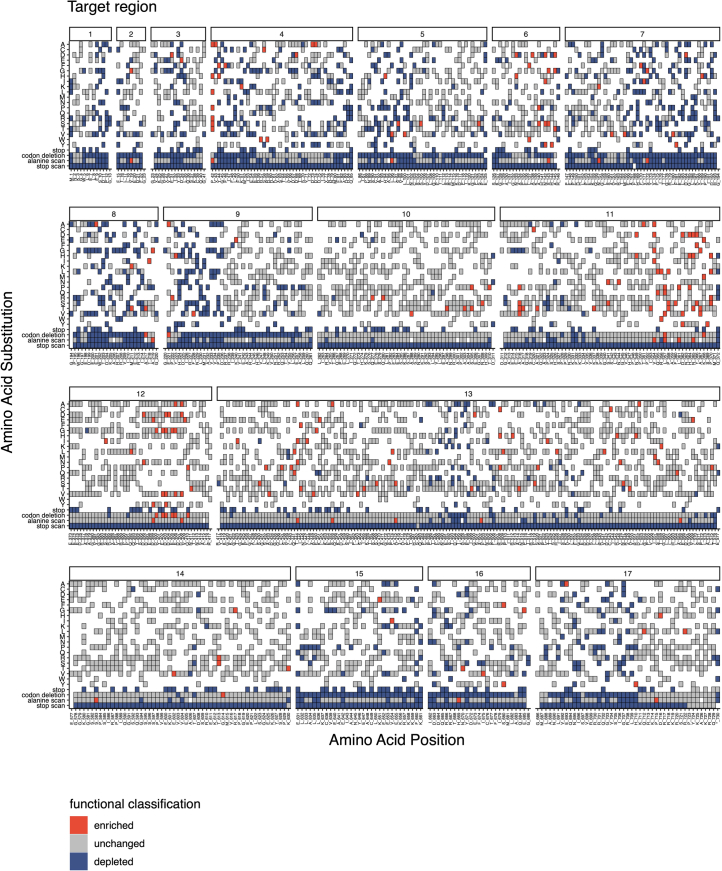
Many variants that we inherit from our parents or acquire de novo or somatically are rare, limiting the precision with which we can associate them with disease. We performed exhaustive saturation genome editing (SGE) of BAP1, the disruption of which is linked to tumorigenesis and altered neurodevelopment. We experimentally characterized 18,108 unique variants, of which 6,196 were found to have abnormal functions, and then used these data to evaluate phenotypic associations in the UK Biobank. We also characterized variants in a large population-ascertained tumor collection, in cancer pedigrees and ClinVar, and explored the behavior of cancer-associated variants compared to that of variants linked to neurodevelopmental phenotypes. Our analyses demonstrated that disruptive germline BAP1 variants were significantly associated with higher circulating levels of the mitogen IGF-1, suggesting a possible pathological mechanism and therapeutic target. Furthermore, we built a variant classifier with >98% sensitivity and specificity and quantify evidence strengths to aid precision variant interpretation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Star P, et al. Germline BAP1-positive patients: the dilemmas of cancer surveillance and a proposed interdisciplinary consensus monitoring strategy. Eur. J. Cancer. 2018;92:48–53. - PubMed
-
- Fennell DA, et al. Rucaparib in patients with BAP1-deficient or BRCA1-deficient mesothelioma (MiST1): an open-label, single-arm, phase 2a clinical trial. Lancet Respir. Med. 2021;9:593–600. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
