

Saturation genome editing maps the functional spectrum of pathogenic VHL alleles
- PMID: 38969834
- PMCID: PMC11250436
- DOI: 10.1038/s41588-024-01800-z
Saturation genome editing maps the functional spectrum of pathogenic VHL alleles
Abstract
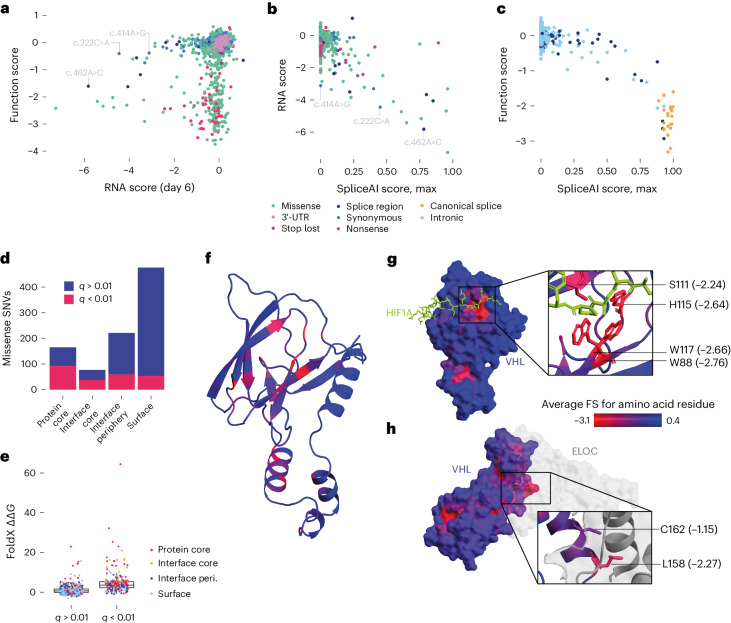
To maximize the impact of precision medicine approaches, it is critical to identify genetic variants underlying disease and to accurately quantify their functional effects. A gene exemplifying the challenge of variant interpretation is the von Hippel-Lindautumor suppressor (VHL). VHL encodes an E3 ubiquitin ligase that regulates the cellular response to hypoxia. Germline pathogenic variants in VHL predispose patients to tumors including clear cell renal cell carcinoma (ccRCC) and pheochromocytoma, and somatic VHL mutations are frequently observed in sporadic renal cancer. Here we optimize and apply saturation genome editing to assay nearly all possible single-nucleotide variants (SNVs) across VHL's coding sequence. To delineate mechanisms, we quantify mRNA dosage effects and compare functional effects in isogenic cell lines. Function scores for 2,268 VHL SNVs identify a core set of pathogenic alleles driving ccRCC with perfect accuracy, inform differential risk across tumor types and reveal new mechanisms by which variants impact function. These results have immediate utility for classifying VHL variants encountered clinically and illustrate how precise functional measurements can resolve pleiotropic and dosage-dependent genotype-phenotype relationships across complete genes.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures















References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
