

Tumor cell-intrinsic MELK enhanced CCL2-dependent immunosuppression to exacerbate hepatocarcinogenesis and confer resistance of HCC to radiotherapy
- PMID: 38970074
- PMCID: PMC11225310
- DOI: 10.1186/s12943-024-02049-0
Tumor cell-intrinsic MELK enhanced CCL2-dependent immunosuppression to exacerbate hepatocarcinogenesis and confer resistance of HCC to radiotherapy
Abstract
Background: The outcome of hepatocellular carcinoma (HCC) is limited by its complex molecular characteristics and changeable tumor microenvironment (TME). Here we focused on elucidating the functional consequences of Maternal embryonic leucine zipper kinase (MELK) in the tumorigenesis, progression and metastasis of HCC, and exploring the effect of MELK on immune cell regulation in the TME, meanwhile clarifying the corresponding signaling networks.
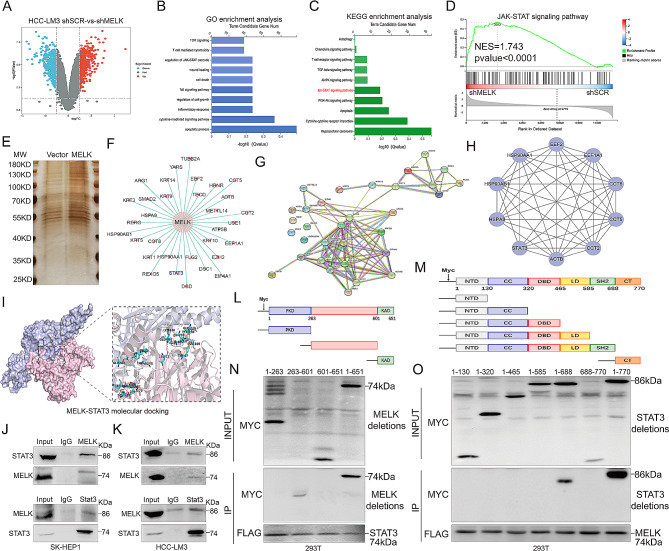
Methods: Bioinformatic analysis was used to validate the prognostic value of MELK for HCC. Murine xenograft assays and HCC lung metastasis mouse model confirmed the role of MELK in tumorigenesis and metastasis in HCC. Luciferase assays, RNA sequencing, immunopurification-mass spectrometry (IP-MS) and coimmunoprecipitation (CoIP) were applied to explore the upstream regulators, downstream essential molecules and corresponding mechanisms of MELK in HCC.
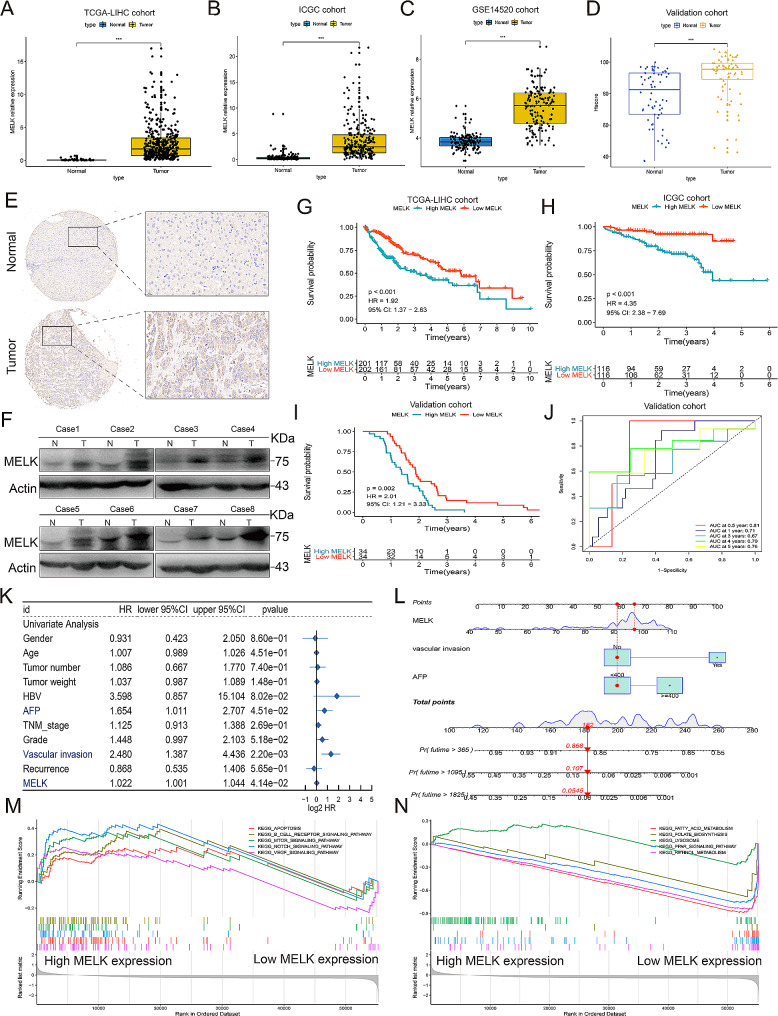
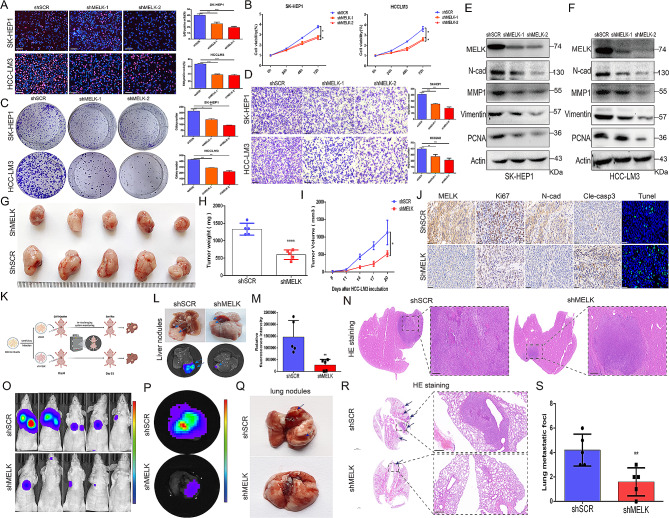
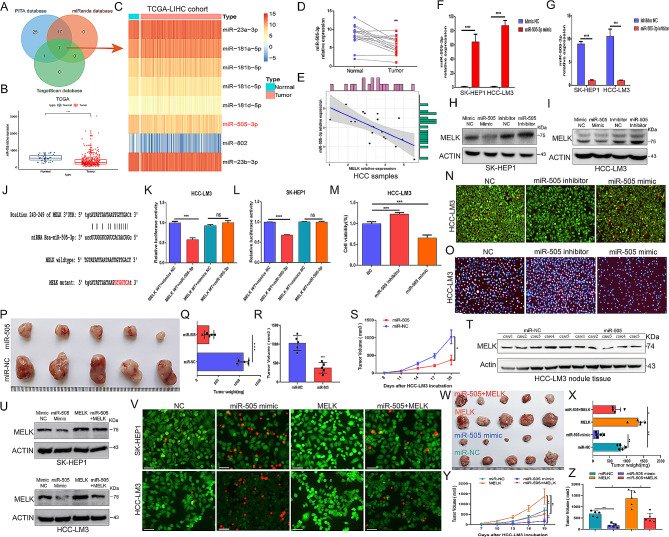
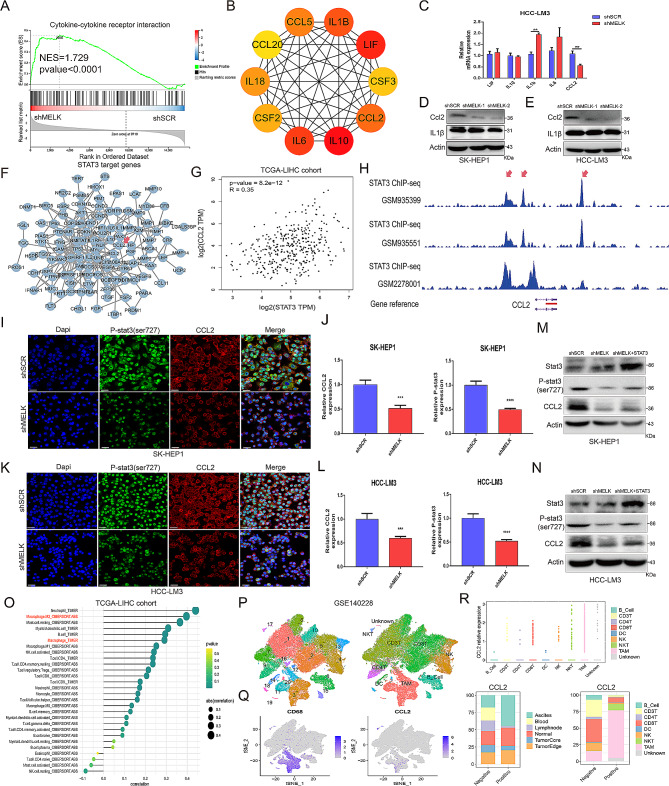
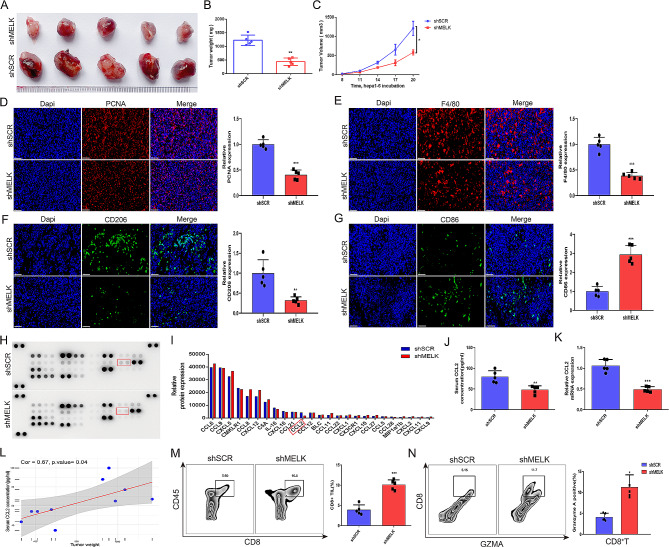
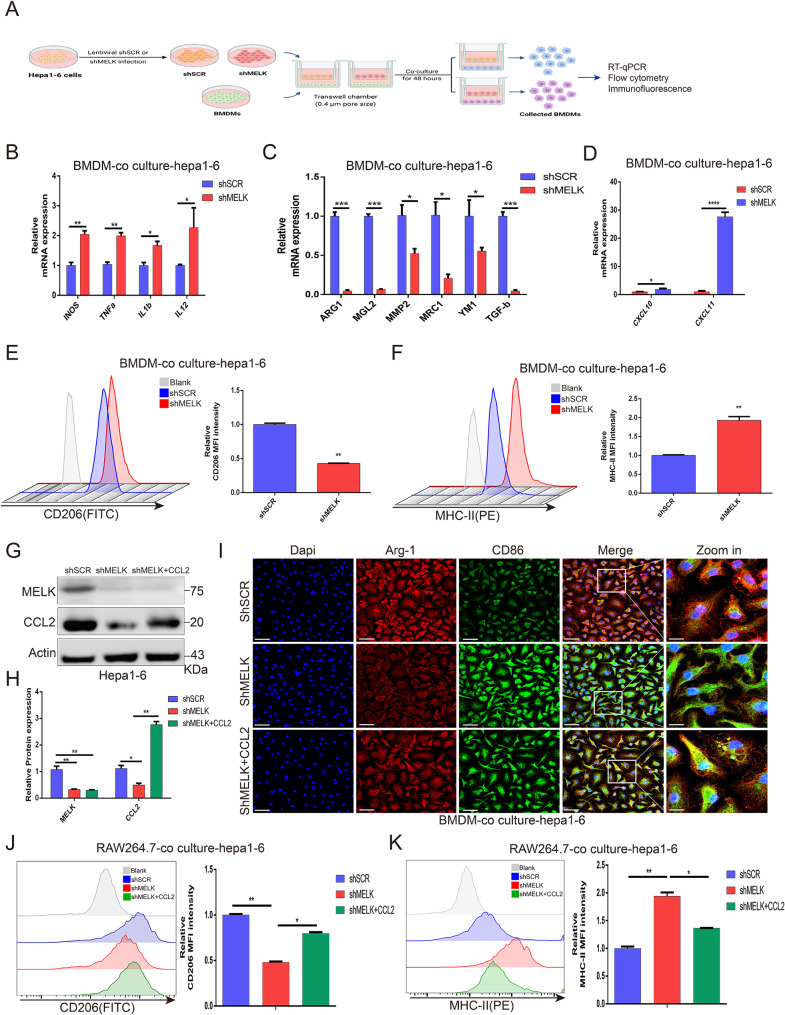
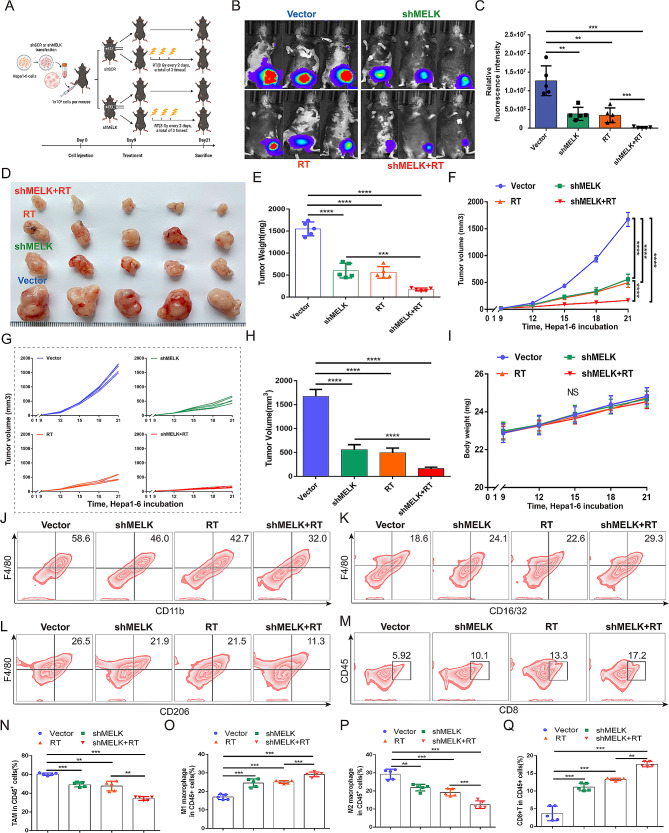
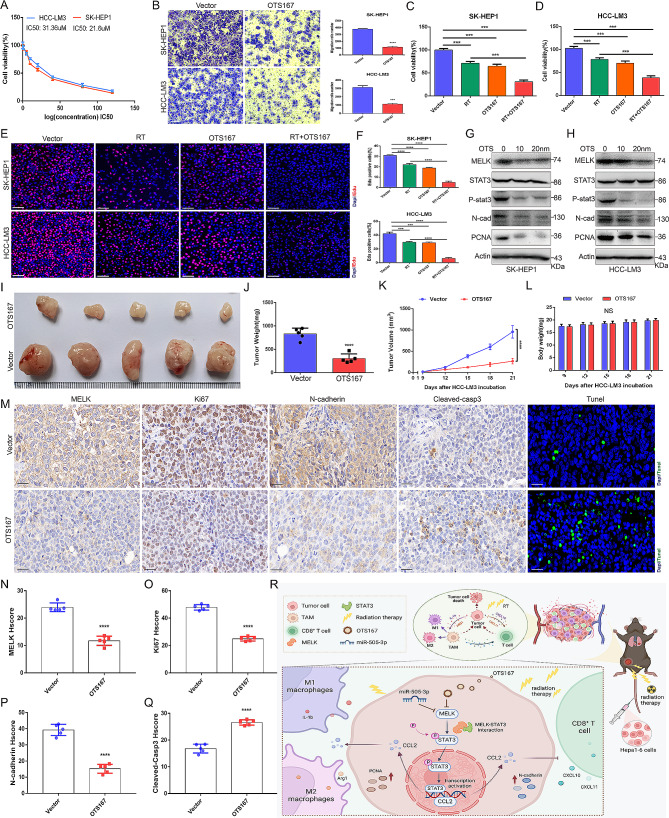
Results: We confirmed MELK to be a reliable prognostic factor of HCC and identified MELK as an effective candidate in facilitating the tumorigenesis, progression, and metastasis of HCC; the effects of MELK depended on the targeted regulation of the upstream factor miR-505-3p and interaction with STAT3, which induced STAT3 phosphorylation and increased the expression of its target gene CCL2 in HCC. In addition, we confirmed that tumor cell-intrinsic MELK inhibition is beneficial in stimulating M1 macrophage polarization, hindering M2 macrophage polarization and inducing CD8 + T-cell recruitment, which are dependent on the alteration of CCL2 expression. Importantly, MELK inhibition amplified RT-related immune effects, thereby synergizing with RT to exert substantial antitumor effects. OTS167, an inhibitor of MELK, was also proven to effectively impair the growth and progression of HCC and exert a superior antitumor effect in combination with radiotherapy (RT).
Conclusions: Altogether, our findings highlight the functional role of MELK as a promising target in molecular therapy and in the combination of RT therapy to improve antitumor effect for HCC.
Keywords: CCL2; Hepatocellular carcinoma (HCC); Radiotherapy (RT); STAT3; Tumor-associated macrophage (TAM); miR-505.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
