

Single-cell resolution characterization of myeloid-derived cell states with implication in cancer outcome
- PMID: 38972873
- PMCID: PMC11228020
- DOI: 10.1038/s41467-024-49916-4
Single-cell resolution characterization of myeloid-derived cell states with implication in cancer outcome
Abstract
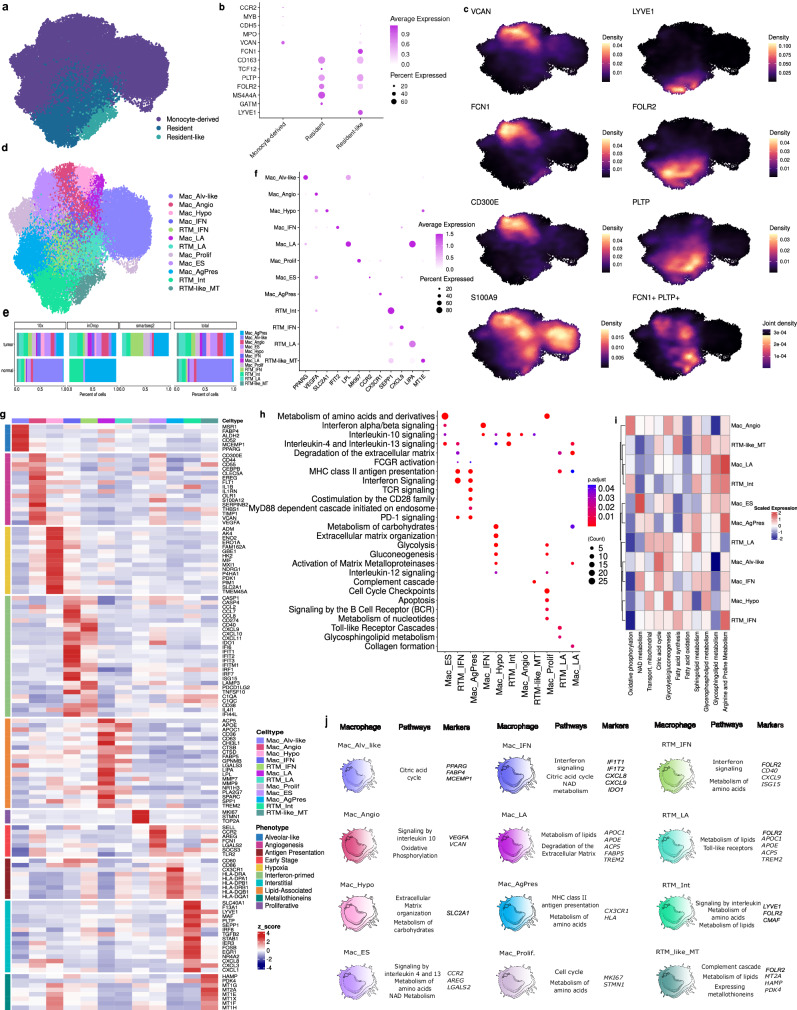
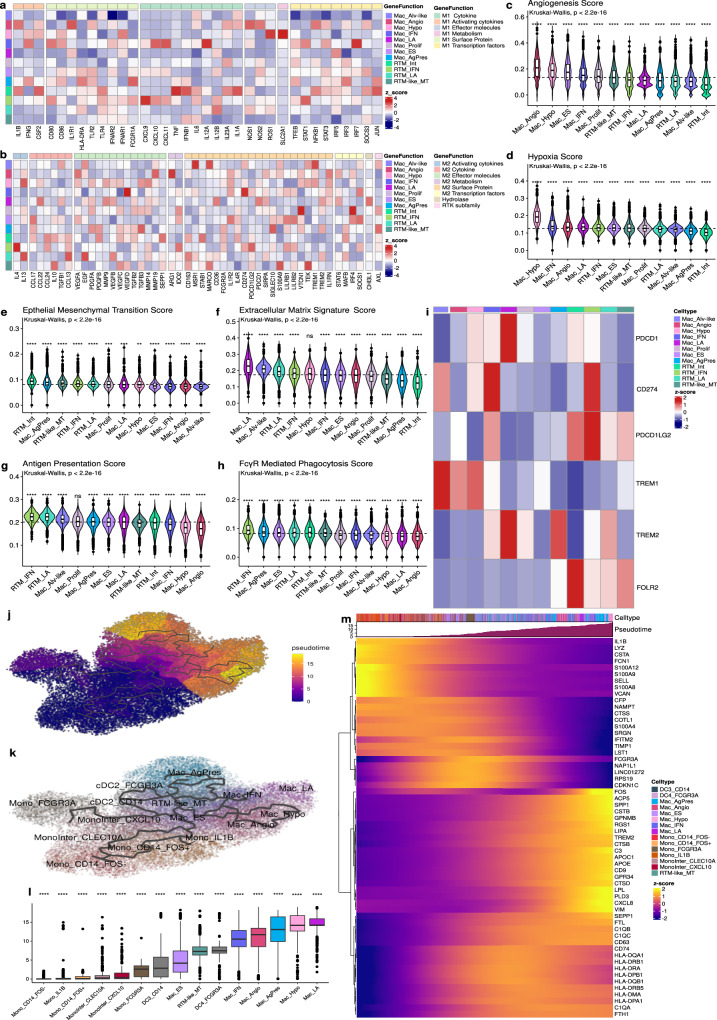
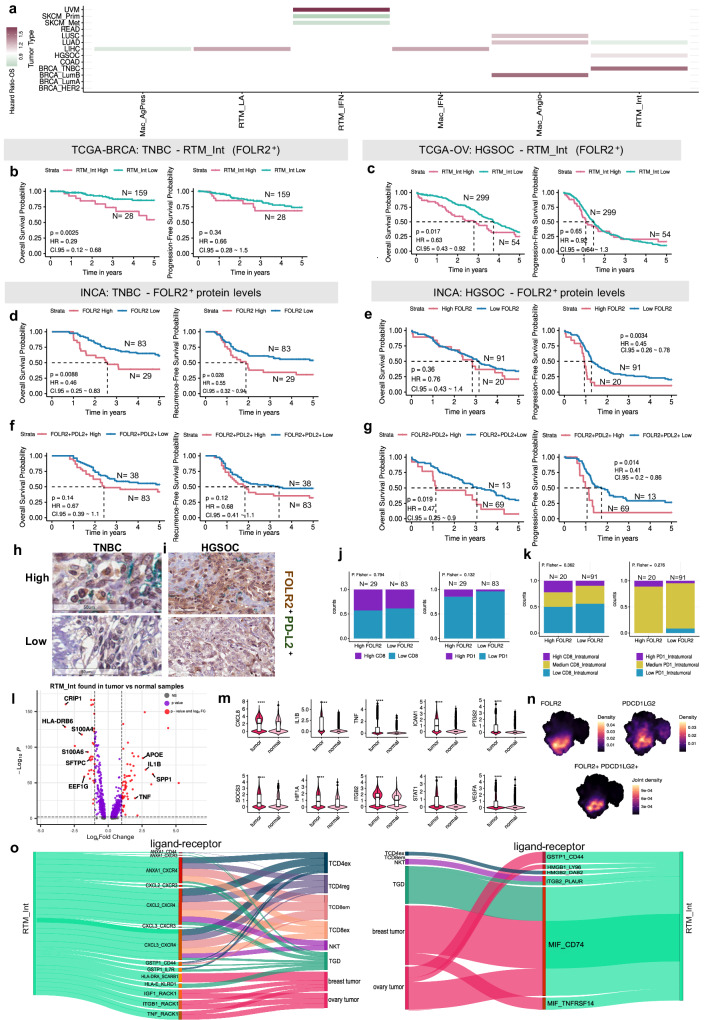
Tumor-associated myeloid-derived cells (MDCs) significantly impact cancer prognosis and treatment responses due to their remarkable plasticity and tumorigenic behaviors. Here, we integrate single-cell RNA-sequencing data from different cancer types, identifying 29 MDC subpopulations within the tumor microenvironment. Our analysis reveals abnormally expanded MDC subpopulations across various tumors and distinguishes cell states that have often been grouped together, such as TREM2+ and FOLR2+ subpopulations. Using deconvolution approaches, we identify five subpopulations as independent prognostic markers, including states co-expressing TREM2 and PD-1, and FOLR2 and PDL-2. Additionally, TREM2 alone does not reliably predict cancer prognosis, as other TREM2+ macrophages show varied associations with prognosis depending on local cues. Validation in independent cohorts confirms that FOLR2-expressing macrophages correlate with poor clinical outcomes in ovarian and triple-negative breast cancers. This comprehensive MDC atlas offers valuable insights and a foundation for futher analyses, advancing strategies for treating solid cancers.
© 2024. The Author(s).
Conflict of interest statement
During the preparation of the manuscript M.A.P. was hired by Egle Therapeutics. The other authors declare no competing interests.
Figures




References
-
- Sharma P, et al. The next decade of immune checkpoint therapy. Cancer Discov. 2021;11:838–857. doi: 10.1158/2159-8290.CD-20-1680. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
