

Epigenetic alterations affecting hematopoietic regulatory networks as drivers of mixed myeloid/lymphoid leukemia
- PMID: 38972954
- PMCID: PMC11228033
- DOI: 10.1038/s41467-024-49811-y
Epigenetic alterations affecting hematopoietic regulatory networks as drivers of mixed myeloid/lymphoid leukemia
Abstract
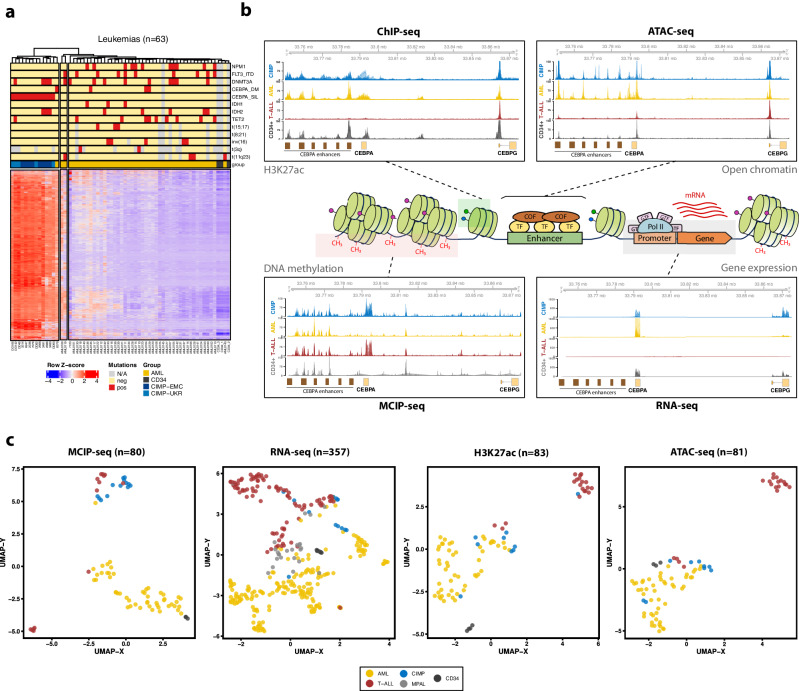
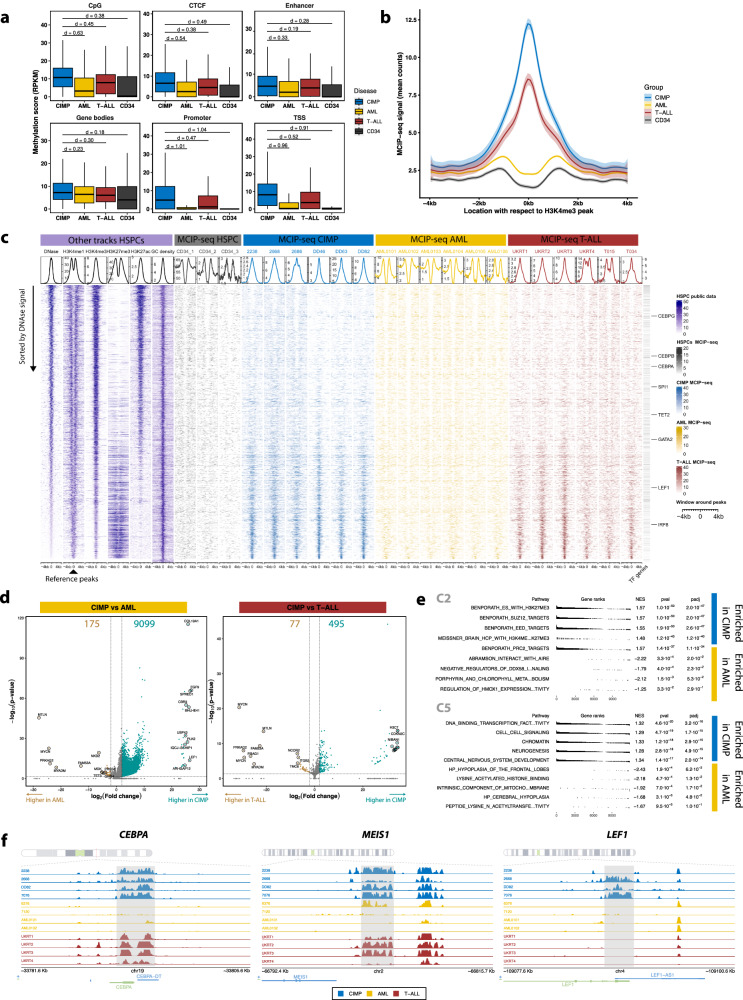
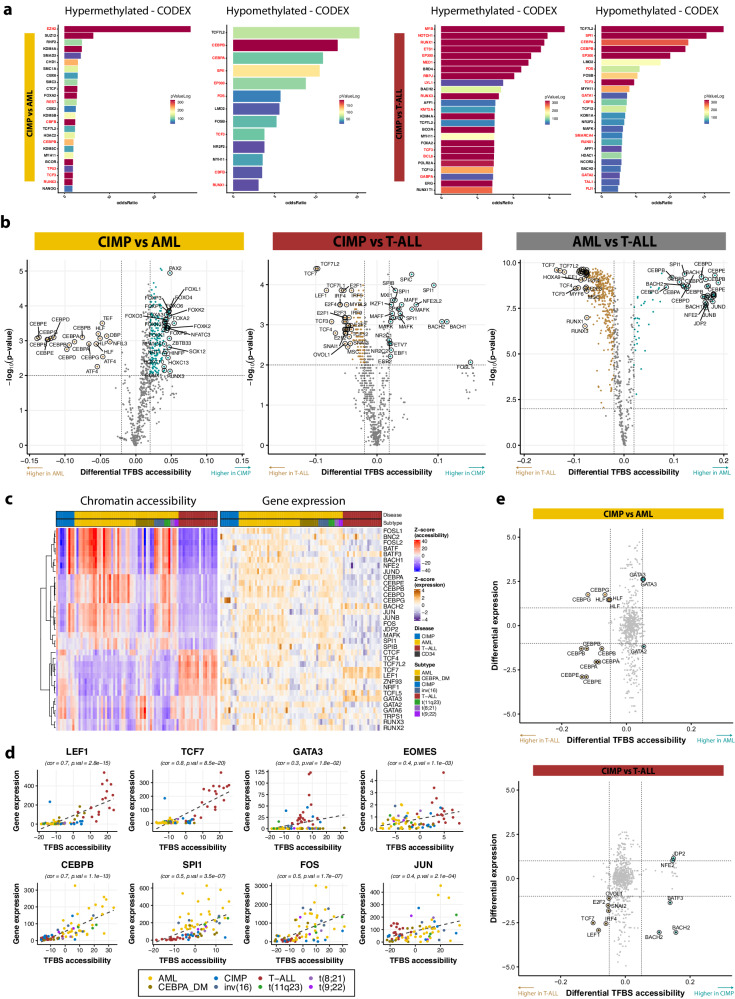
Leukemias with ambiguous lineage comprise several loosely defined entities, often without a clear mechanistic basis. Here, we extensively profile the epigenome and transcriptome of a subgroup of such leukemias with CpG Island Methylator Phenotype. These leukemias exhibit comparable hybrid myeloid/lymphoid epigenetic landscapes, yet heterogeneous genetic alterations, suggesting they are defined by their shared epigenetic profile rather than common genetic lesions. Gene expression enrichment reveals similarity with early T-cell precursor acute lymphoblastic leukemia and a lymphoid progenitor cell of origin. In line with this, integration of differential DNA methylation and gene expression shows widespread silencing of myeloid transcription factors. Moreover, binding sites for hematopoietic transcription factors, including CEBPA, SPI1 and LEF1, are uniquely inaccessible in these leukemias. Hypermethylation also results in loss of CTCF binding, accompanied by changes in chromatin interactions involving key transcription factors. In conclusion, epigenetic dysregulation, and not genetic lesions, explains the mixed phenotype of this group of leukemias with ambiguous lineage. The data collected here constitute a useful and comprehensive epigenomic reference for subsequent studies of acute myeloid leukemias, T-cell acute lymphoblastic leukemias and mixed-phenotype leukemias.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Genome-wide epigenetic analysis delineates a biologically distinct immature acute leukemia with myeloid/T-lymphoid features.Blood. 2009 Mar 19;113(12):2795-804. doi: 10.1182/blood-2008-08-172387. Epub 2009 Jan 23. Blood. 2009. PMID: 19168792 Free PMC article.
-
14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia.Blood. 2021 Sep 2;138(9):773-784. doi: 10.1182/blood.2020010510. Blood. 2021. PMID: 33876209 Free PMC article. Clinical Trial.
-
Epigenetic control of SPI1 gene by CTCF and ISWI ATPase SMARCA5.PLoS One. 2014 Feb 3;9(2):e87448. doi: 10.1371/journal.pone.0087448. eCollection 2014. PLoS One. 2014. PMID: 24498324 Free PMC article.
-
Targeting epigenetic programs in MLL-rearranged leukemias.Hematology Am Soc Hematol Educ Program. 2011;2011:354-60. doi: 10.1182/asheducation-2011.1.354. Hematology Am Soc Hematol Educ Program. 2011. PMID: 22160057 Review.
-
DNA Methylation in T-Cell Acute Lymphoblastic Leukemia: In Search for Clinical and Biological Meaning.Int J Mol Sci. 2021 Jan 30;22(3):1388. doi: 10.3390/ijms22031388. Int J Mol Sci. 2021. PMID: 33573325 Free PMC article. Review.
Cited by
-
Multiomics profiles of genome-wide alterations in H3K27ac in different lung lobes after acute graft-versus-host disease with MSCs treatment.Front Immunol. 2025 May 15;16:1570916. doi: 10.3389/fimmu.2025.1570916. eCollection 2025. Front Immunol. 2025. PMID: 40443676 Free PMC article.
-
Transcriptional profiling directs the classification of acute leukemias of ambiguous lineage into AML, B-ALL, or T-ALL.Hemasphere. 2025 Aug 19;9(8):e70195. doi: 10.1002/hem3.70195. eCollection 2025 Aug. Hemasphere. 2025. PMID: 40838101 Free PMC article.
-
Epigenetic regulation of MED12: a key contributor to the leukemic chromatin landscape and transcriptional dysregulation.Epigenetics Chromatin. 2025 Jul 14;18(1):44. doi: 10.1186/s13072-025-00610-9. Epigenetics Chromatin. 2025. PMID: 40660382 Free PMC article.
-
Liquid Biopsy and Epigenetic Signatures in AML, ALL, and CNS Tumors: Diagnostic and Monitoring Perspectives.Int J Mol Sci. 2025 Aug 5;26(15):7547. doi: 10.3390/ijms26157547. Int J Mol Sci. 2025. PMID: 40806675 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
