

Insights and Challenges in Correcting Force Field Based Solvation Free Energies Using a Neural Network Potential
- PMID: 38976601
- PMCID: PMC11264272
- DOI: 10.1021/acs.jpcb.4c01417
Insights and Challenges in Correcting Force Field Based Solvation Free Energies Using a Neural Network Potential
Abstract
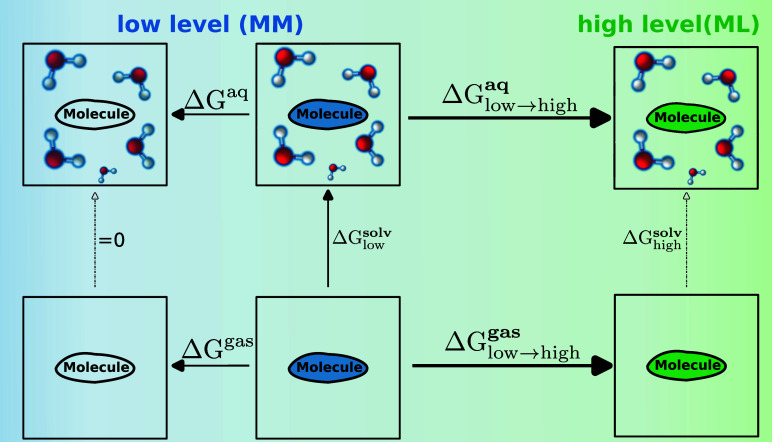
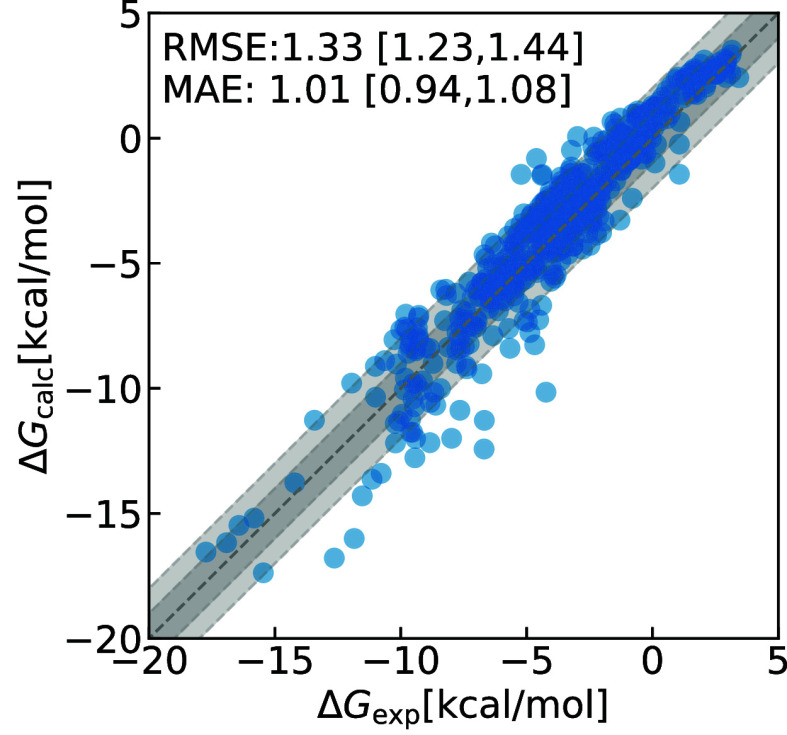
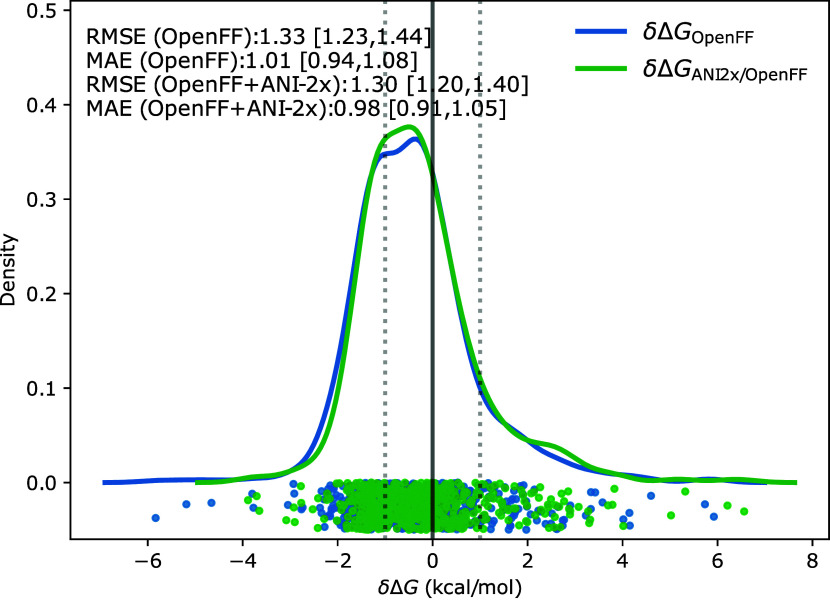
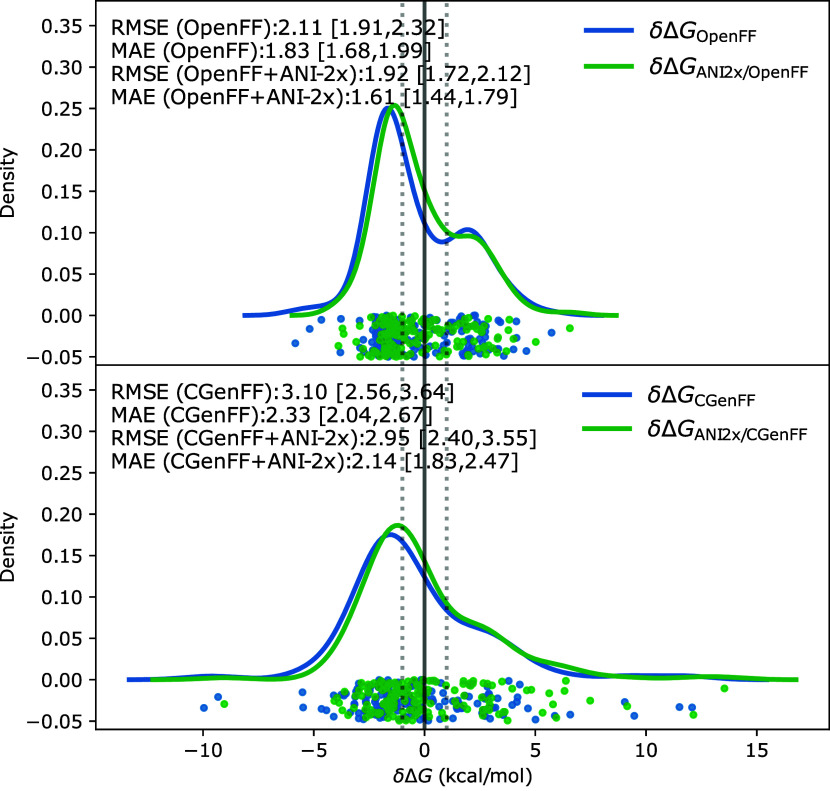
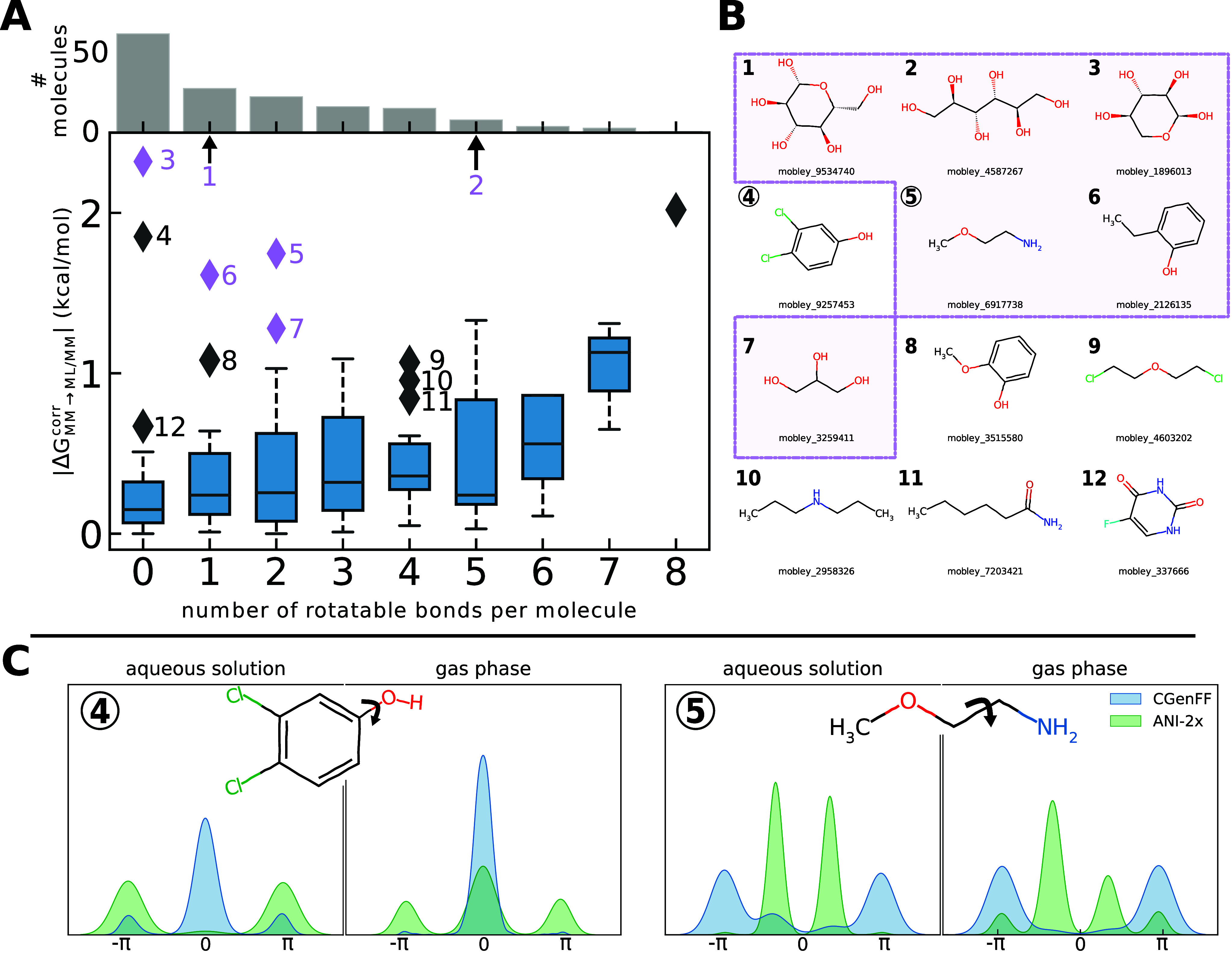
We present a comprehensive study investigating the potential gain in accuracy for calculating absolute solvation free energies (ASFE) using a neural network potential to describe the intramolecular energy of the solute. We calculated the ASFE for most compounds from the FreeSolv database using the Open Force Field (OpenFF) and compared them to earlier results obtained with the CHARMM General Force Field (CGenFF). By applying a nonequilibrium (NEQ) switching approach between the molecular mechanics (MM) description (either OpenFF or CGenFF) and the neural net potential (NNP)/MM level of theory (using ANI-2x as the NNP potential), we attempted to improve the accuracy of the calculated ASFEs. The predictive performance of the results did not change when this approach was applied to all 589 small molecules in the FreeSolv database that ANI-2x can describe. When selecting a subset of 156 molecules, focusing on compounds where the force fields performed poorly, we saw a slight improvement in the root-mean-square error (RMSE) and mean absolute error (MAE). The majority of our calculations utilized unidirectional NEQ protocols based on Jarzynski's equation. Additionally, we conducted bidirectional NEQ switching for a subset of 156 solutes. Notably, only a small fraction (10 out of 156) exhibited statistically significant discrepancies between unidirectional and bidirectional NEQ switching free energy estimates.
Conflict of interest statement
The authors declare the following competing financial interest(s): S.B. is a consultant for Exscientia.
Figures
Similar articles
-
Benchmarking Force Field and the ANI Neural Network Potentials for the Torsional Potential Energy Surface of Biaryl Drug Fragments.J Chem Inf Model. 2020 Dec 28;60(12):6258-6268. doi: 10.1021/acs.jcim.0c00904. Epub 2020 Dec 2. J Chem Inf Model. 2020. PMID: 33263401
-
Predicting Solvation Free Energies from the Minnesota Solvation Database Using Classical Density Functional Theory Based on the PC-SAFT Equation of State.J Phys Chem B. 2024 Apr 18;128(15):3677-3688. doi: 10.1021/acs.jpcb.3c07447. Epub 2024 Apr 5. J Phys Chem B. 2024. PMID: 38579126
-
Reweighting from Molecular Mechanics Force Fields to the ANI-2x Neural Network Potential.J Chem Theory Comput. 2024 Apr 9;20(7):2719-2728. doi: 10.1021/acs.jctc.3c01274. Epub 2024 Mar 25. J Chem Theory Comput. 2024. PMID: 38527958
-
Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?J Comput Aided Mol Des. 2019 Feb;33(2):133-203. doi: 10.1007/s10822-018-0111-4. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506158 Review.
-
Accurate Prediction of Aqueous Free Solvation Energies Using 3D Atomic Feature-Based Graph Neural Network with Transfer Learning.J Chem Inf Model. 2022 Apr 25;62(8):1840-1848. doi: 10.1021/acs.jcim.2c00260. Epub 2022 Apr 14. J Chem Inf Model. 2022. PMID: 35422122 Free PMC article. Review.
Cited by
-
ABCG2: A Milestone Charge Model for Accurate Solvation Free Energy Calculation.J Chem Theory Comput. 2025 Mar 25;21(6):3032-3043. doi: 10.1021/acs.jctc.5c00038. Epub 2025 Mar 11. J Chem Theory Comput. 2025. PMID: 40068154 Free PMC article.
-
Quantification of the Impact of Structure Quality on Predicted Binding Free Energy Accuracy.J Chem Inf Model. 2025 Jul 14;65(13):6927-6938. doi: 10.1021/acs.jcim.5c00947. Epub 2025 Jun 29. J Chem Inf Model. 2025. PMID: 40583448 Free PMC article.
References
-
- Cournia Z.; Chipot C.; Roux B.; York D. M.; Sherman W.. Free Energy Methods in Drug Discovery: Current State and Future Directions; American Chemical Society, 2021; pp 1–38.
Grants and funding
LinkOut - more resources
Full Text Sources
