

Origins of High-Activity Cage-Catalyzed Michael Addition
- PMID: 38976816
- PMCID: PMC11258793
- DOI: 10.1021/jacs.4c05160
Origins of High-Activity Cage-Catalyzed Michael Addition
Abstract
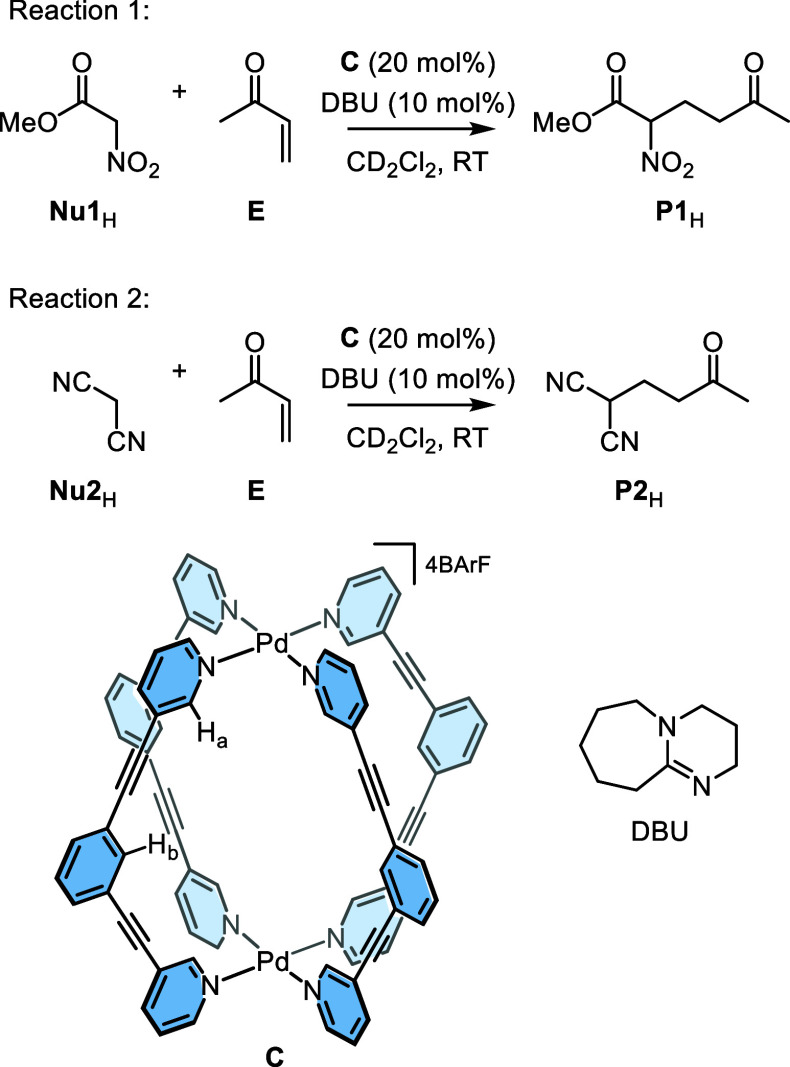
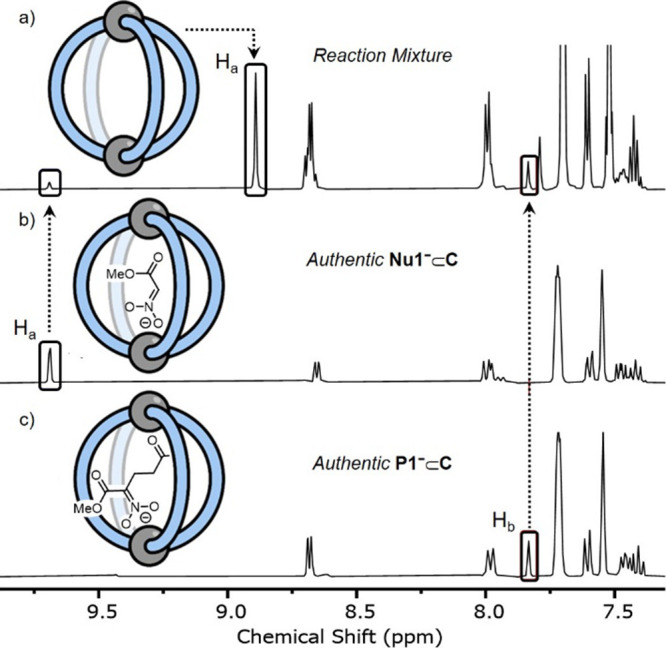
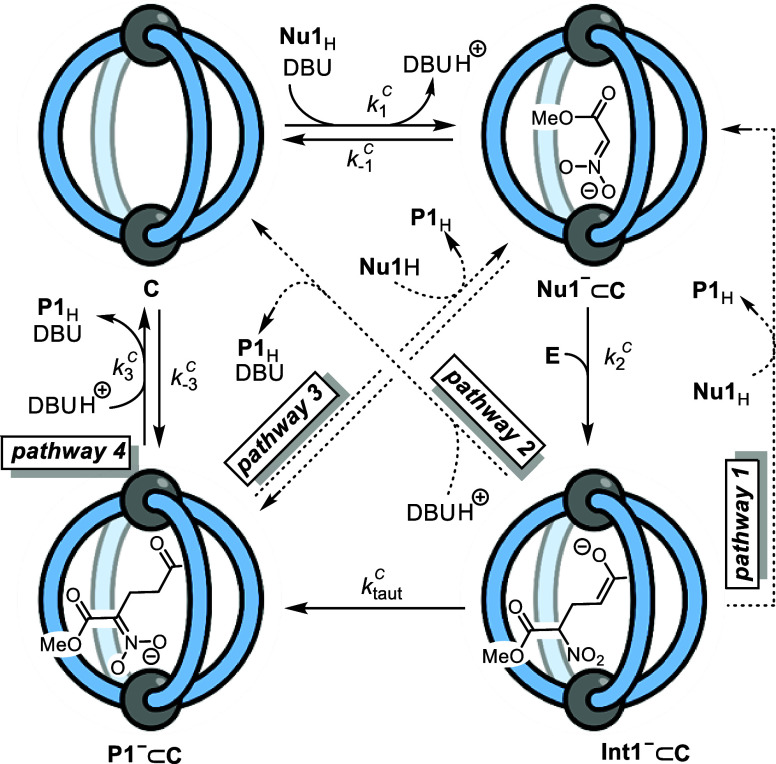
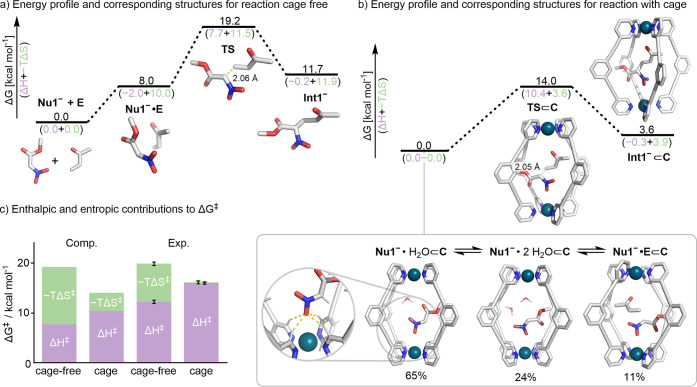
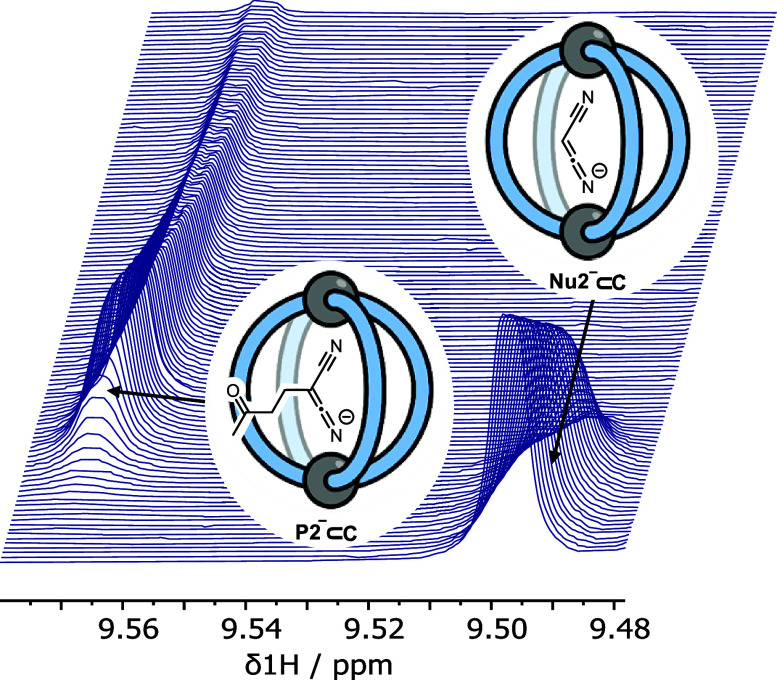
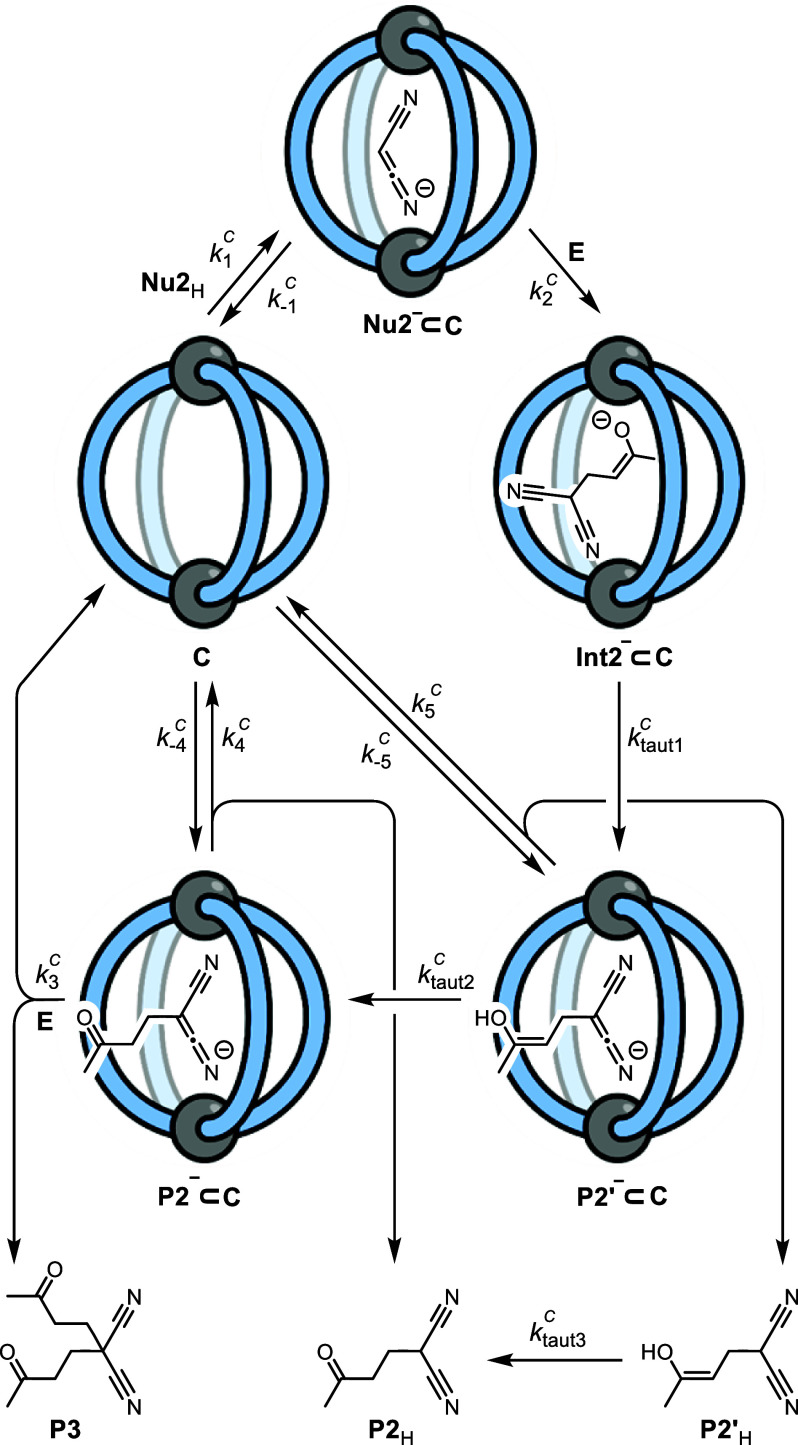
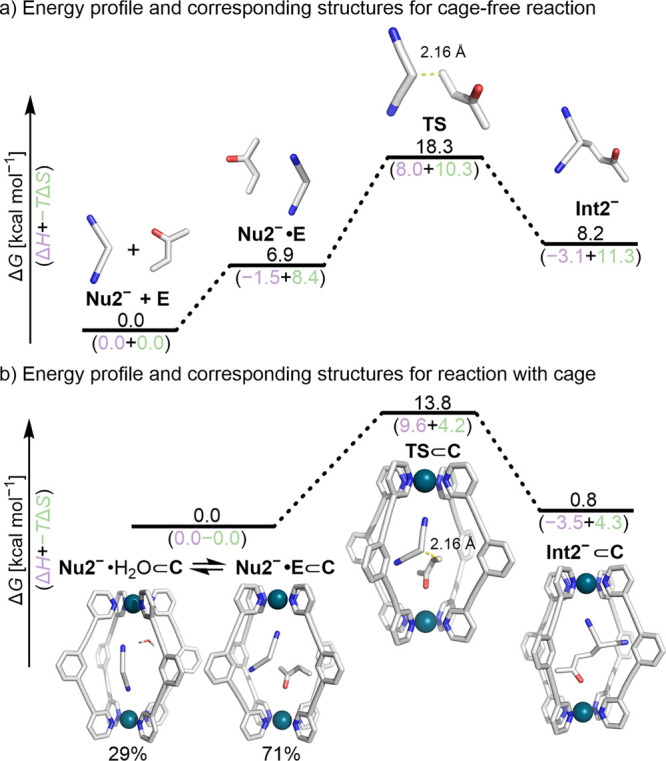
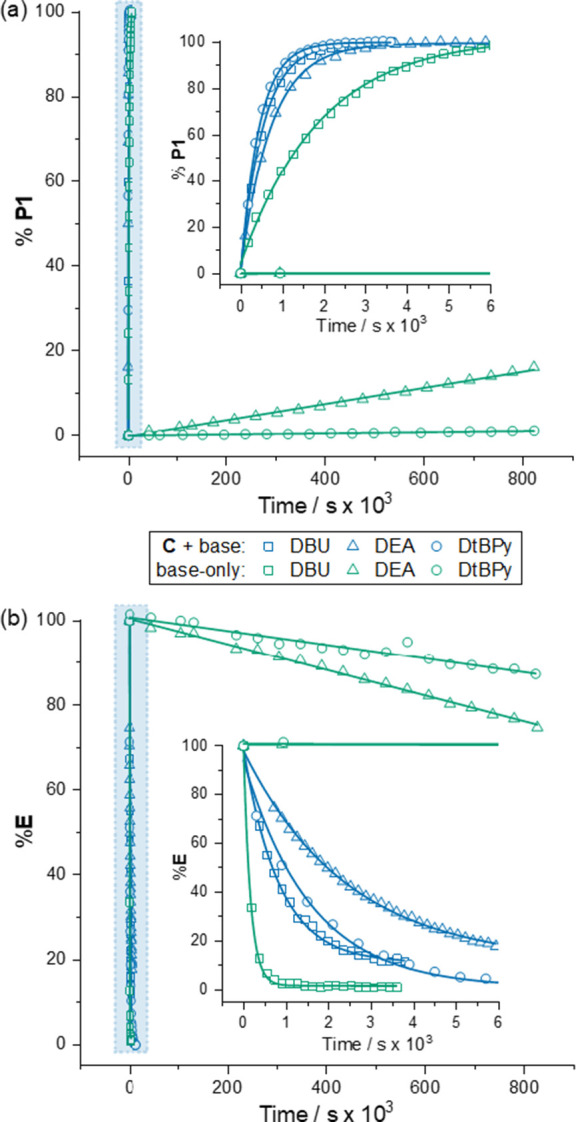
Cage catalysis continues to create significant interest, yet catalyst function remains poorly understood. Herein, we report mechanistic insights into coordination-cage-catalyzed Michael addition using kinetic and computational methods. The study has been enabled by the detection of identifiable catalyst intermediates, which allow the evolution of different cage species to be monitored and modeled alongside reactants and products. The investigations show that the overall acceleration results from two distinct effects. First, the cage reaction shows a thousand-fold increase in the rate constant for the turnover-limiting C-C bond-forming step compared to a reference state. Computational modeling and experimental analysis of activation parameters indicate that this stems from a significant reduction in entropy, suggesting substrate coencapsulation. Second, the cage markedly acidifies the bound pronucleophile, shifting this equilibrium by up to 6 orders of magnitude. The combination of these two factors results in accelerations up to 109 relative to bulk-phase reference reactions. We also show that the catalyst can fundamentally alter the reaction mechanism, leading to intermediates and products that are not observable outside of the cage. Collectively, the results show that cage catalysis can proceed with very high activity and unique selectivity by harnessing a series of individually weak noncovalent interactions.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Rideout D. C.; Breslow R. Hydrophobic acceleration of Diels-Alder reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. 10.1021/ja00546a048. - DOI
-
- Mackay L. G.; Wylie R. S.; Sanders J. K. M. Catalytic Acyl Transfer by a Cyclic Porphyrin Trimer: Efficient Turnover without Product Inhibition. J. Am. Chem. Soc. 1994, 116, 3141–3142. 10.1021/ja00086a061. - DOI
-
- Tehrani F. N.; Assaf K. I.; Hein R.; Jensen C. M. E.; Nugent T. C.; Nau W. M. Supramolecular Catalysis of a Catalysis-Resistant Diels–Alder Reaction: Almost Theoretical Acceleration of Cyclopentadiene Dimerization inside Cucurbit[7]uril. ACS Catal. 2022, 12, 2261–2269. 10.1021/acscatal.1c05659. - DOI
LinkOut - more resources
Full Text Sources
