

This is a preprint.
RNAi-mediated silencing of SOD1 profoundly extends survival and functional outcomes in ALS mice
- PMID: 38979291
- PMCID: PMC11230209
- DOI: 10.1101/2024.06.20.599943
RNAi-mediated silencing of SOD1 profoundly extends survival and functional outcomes in ALS mice
Update in
-
RNAi-mediated silencing of SOD1 profoundly extends survival and functional outcomes in ALS mice.Mol Ther. 2025 Aug 6;33(8):3917-3938. doi: 10.1016/j.ymthe.2025.05.010. Epub 2025 May 9. Mol Ther. 2025. PMID: 40349108
Abstract
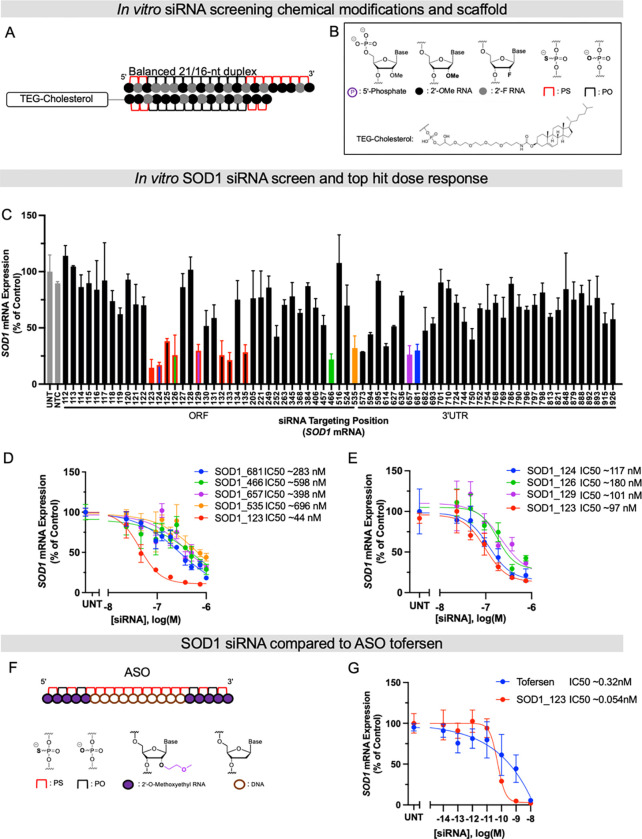
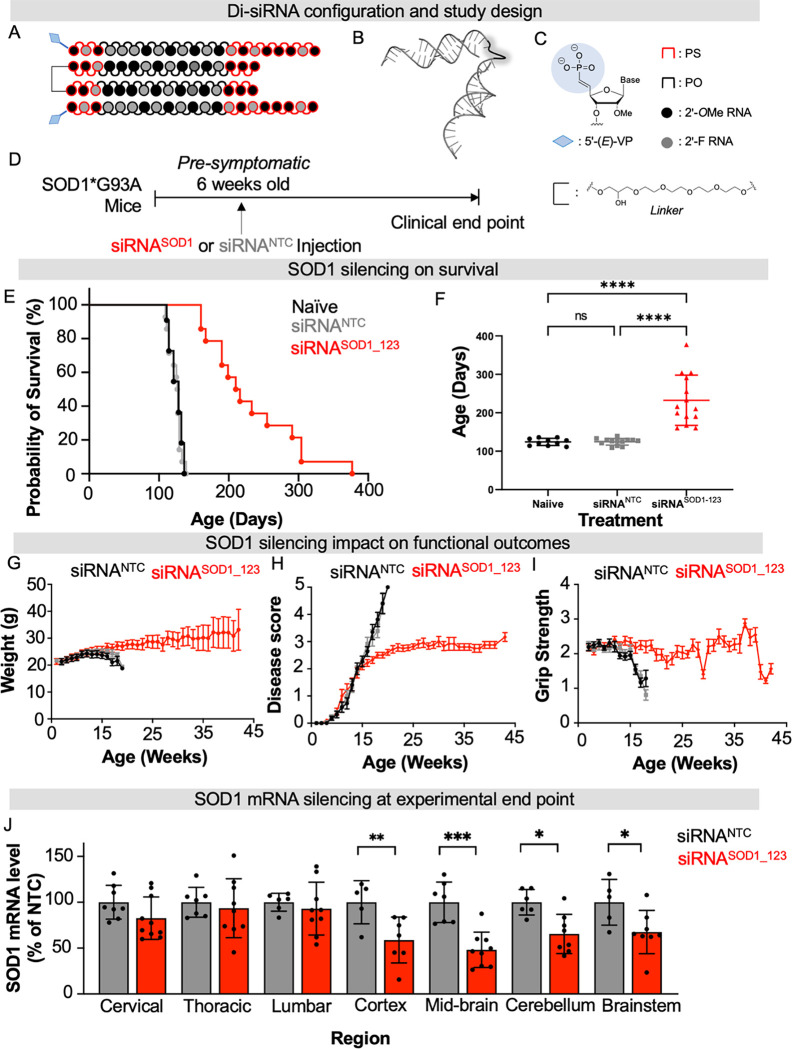
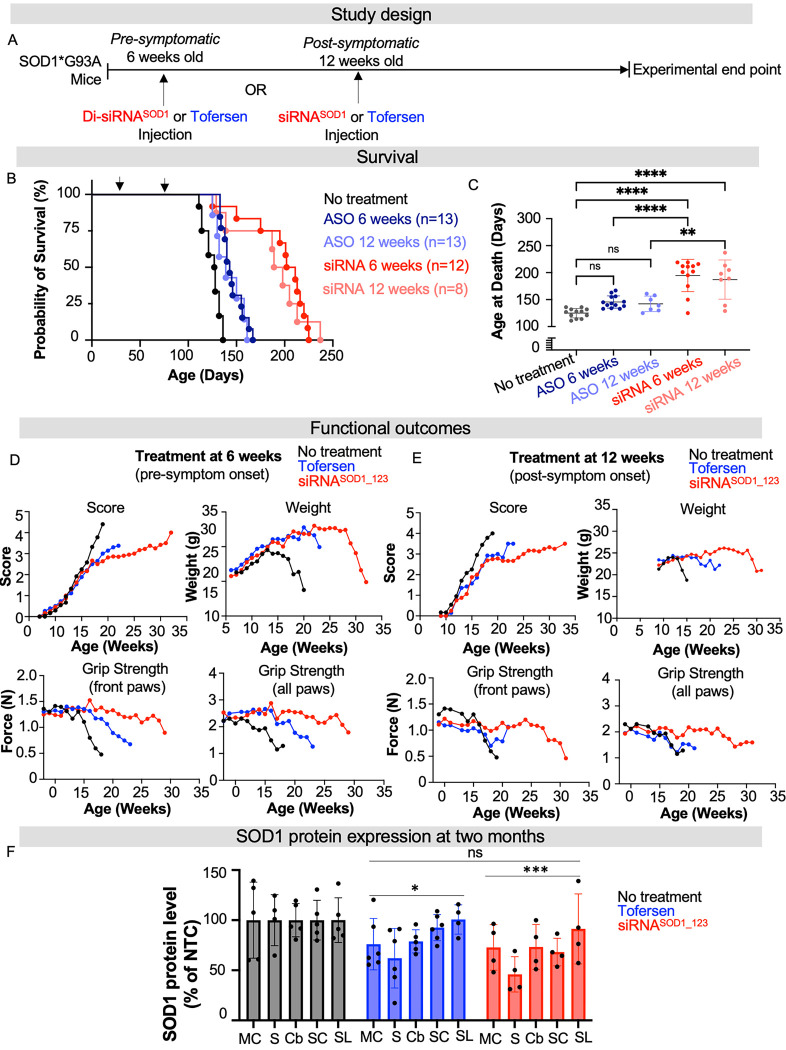
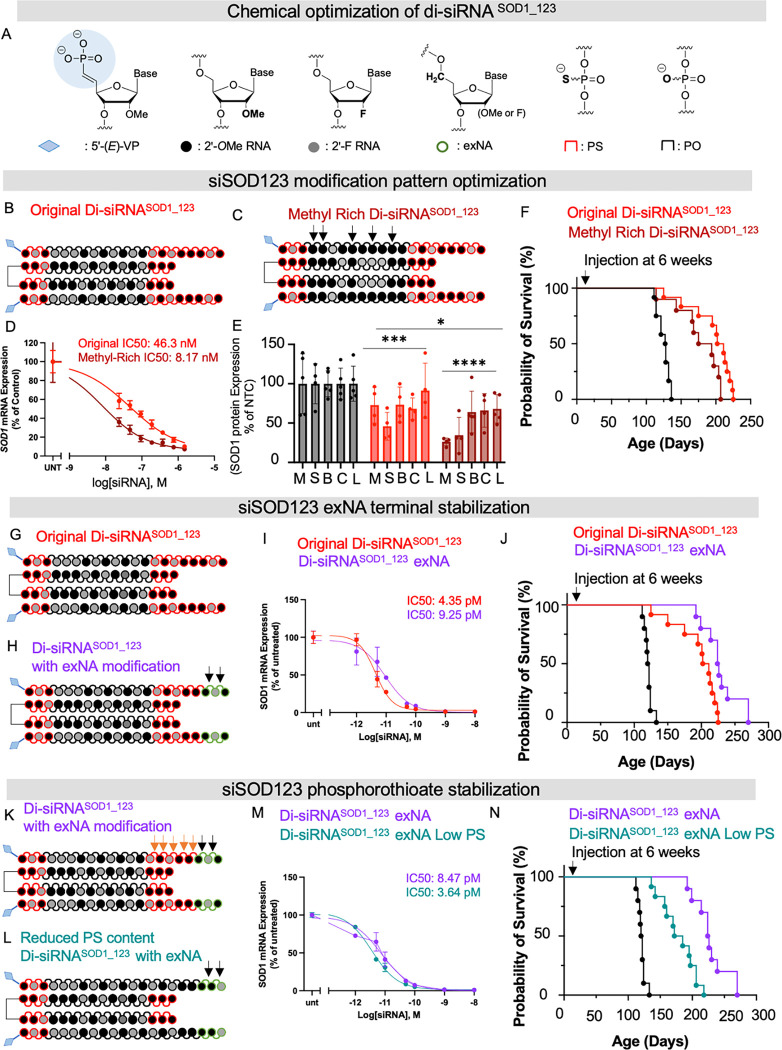
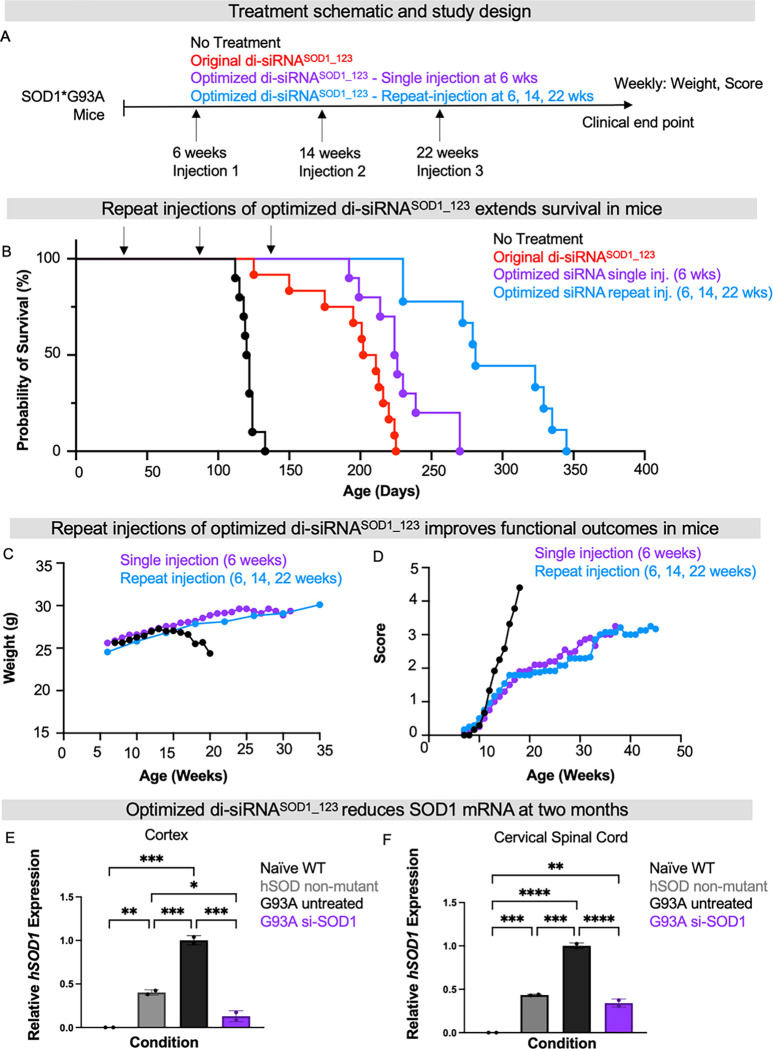
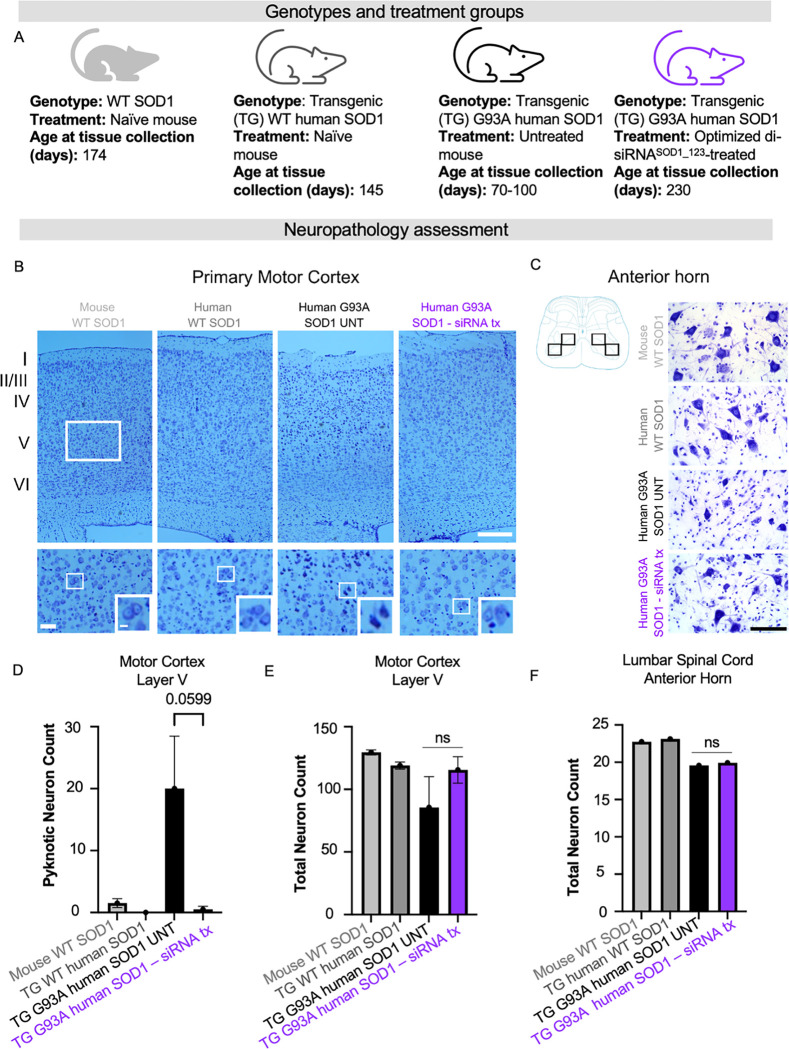
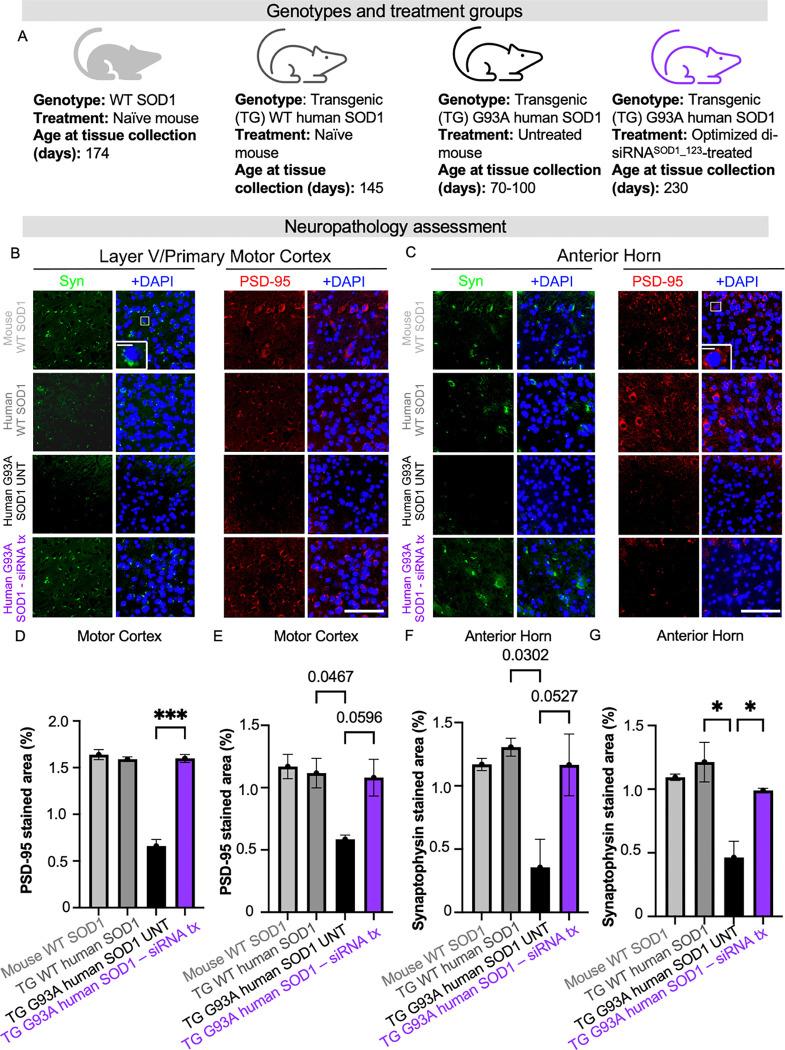
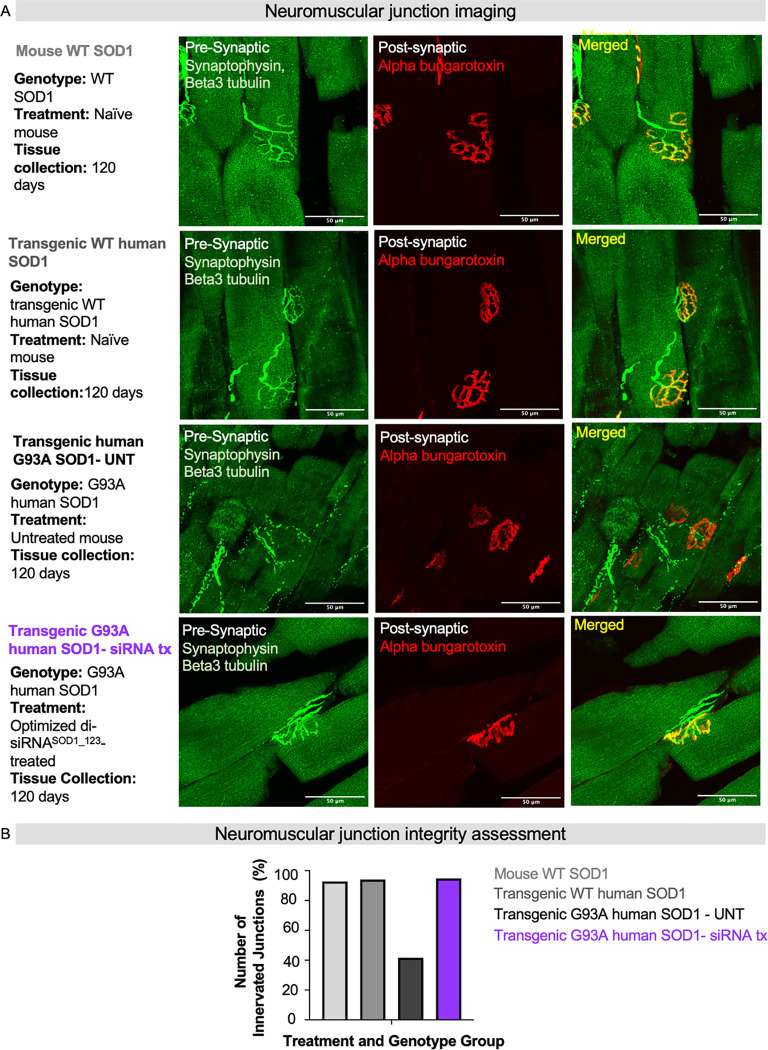
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative condition, with 20% of familial and 2-3% of sporadic cases linked to mutations in the cytosolic superoxide dismutase (SOD1) gene. Mutant SOD1 protein is toxic to motor neurons, making SOD1 gene lowering a promising approach, supported by preclinical data and the 2023 FDA approval of the GapmeR ASO targeting SOD1, tofersen. Despite the approval of an ASO and the optimism it brings to the field, the pharmacodynamics and pharmacokinetics of therapeutic SOD1 modulation can be improved. Here, we developed a chemically stabilized divalent siRNA scaffold (di-siRNA) that effectively suppresses SOD1 expression in vitro and in vivo. With optimized chemical modification, it achieves remarkable CNS tissue permeation and SOD1 silencing in vivo. Administered intraventricularly, di-siRNASOD1 extended survival in SOD1-G93A ALS mice, surpassing survival previously seen in these mice by ASO modalities, slowed disease progression, and prevented ALS neuropathology. These properties offer an improved therapeutic strategy for SOD1-mediated ALS and may extend to other dominantly inherited neurological disorders.
Conflict of interest statement
Competing interests: AK is a co-founder, on the scientific advisory board, and holds equities of Atalanta Therapeutics; AK is a founder of Comanche Pharmaceuticals, and on the scientific advisory board of Aldena Therapeutics, AlltRNA, Prime Medicine, and EVOX Therapeutics; Select authors hold patents or on patent applications relating to the divalent siRNA, SOD1-targeting oligonucleotides, and the methods described in this report. NH received compensation for other services from Myrobalan Inc. and General Dynamics during the conduct of this study, unrelated to the present work.
Figures
References
-
- Talbott E. O., Malek A. M., Lacomis D., The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol 138, 225–238 (2016). - PubMed
-
- Zhang S., Cooper-Knock J., Weimer A. K., Shi M., Moll T., Marshall J. N. G., Harvey C., Nezhad H. G., Franklin J., Souza C. D. S., Ning K., Wang C., Li J., Dilliott A. A., Farhan S., Elhaik E., Pasniceanu I., Livesey M. R., Eitan C., Hornstein E., Kenna K. P., Veldink J. H., Ferraiuolo L., Shaw P. J., Snyder M. P., Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 110, 992–1008.e1011 (2022). - PMC - PubMed
-
- van Rheenen W., van der Spek R. A. A., Bakker M. K., van Vugt J. J. F. A., Hop P. J., Zwamborn R. A. J., de Klein N., Westra H.-J., Bakker O. B., Deelen P., Shireby G., Hannon E., Moisse M., Baird D., Restuadi R., Dolzhenko E., Dekker A. M., Gawor K., Westeneng H.-J., Tazelaar G. H. P., van Eijk K. R., Kooyman M., Byrne R. P., Doherty M., Heverin M., Al Khleifat A., Iacoangeli A., Shatunov A., Ticozzi N., Cooper-Knock J., Smith B. N., Gromicho M., Chandran S., Pal S., Morrison K. E., Shaw P. J., Hardy J., Orrell R. W., Sendtner M., Meyer T., Başak N., van der Kooi A. J., Ratti A., Fogh I., Gellera C., Lauria G., Corti S., Cereda C., Sproviero D., D’Alfonso S., Sorarù G., Siciliano G., Filosto M., Padovani A., Chiò A., Calvo A., Moglia C., Brunetti M., Canosa A., Grassano M., Beghi E., Pupillo E., Logroscino G., Nefussy B., Osmanovic A., Nordin A., Lerner Y., Zabari M., Gotkine M., Baloh R. H., Bell S., Vourc’h P., Corcia P., Couratier P., Millecamps S., Meininger V., Salachas F., Mora Pardina J. S., Assialioui A., Rojas-García R., Dion P. A., Ross J. P., Ludolph A. C., Weishaupt J. H., Brenner D., Freischmidt A., Bensimon G., Brice A., Durr A., Payan C. A. M., Saker-Delye S., Wood N. W., Topp S., Rademakers R., Tittmann L., Lieb W., Franke A., Ripke S., Braun A., Kraft J., Whiteman D. C., Olsen C. M., Uitterlinden A. G., Hofman A., Rietschel M., Cichon S., Nöthen M. M., Amouyel P., Comi G., Riva N., Lunetta C., Gerardi F., Cotelli M. S., Rinaldi F., Chiveri L., Guaita M. C., Perrone P., Ceroni M., Diamanti L., Ferrarese C., Tremolizzo L., Delodovici M. L., Bono G., Canosa A., Manera U., Vasta R., Bombaci A., Casale F., Fuda G., Salamone P., Iazzolino B., Peotta L., Cugnasco P., De Marco G., Torrieri M. C., Palumbo F., Gallone S., Barberis M., Sbaiz L., Gentile S., Mauro A., Mazzini L., De Marchi F., Corrado L., D’Alfonso S., Bertolotto A., Gionco M., Leotta D., Odddenino E., Imperiale D., Cavallo R., Pignatta P., De Mattei M., Geda C., Papurello D. M., Gusmaroli G., Comi C., Labate C., Ruiz L., Ferrandi D., Rota E., Aguggia M., Di Vito N., Meineri P., Ghiglione P., Launaro N., Dotta M., Di Sapio A., Giardini G., Tiloca C., Peverelli S., Taroni F., Pensato V., Castellotti B., Comi G. P., Del Bo R., Ceroni M., Gagliardi S., Corrado L., Mazzini L., Raggi F., Simoncini C., Lo Gerfo A., Inghilleri M., Ferlini A., Simone I. L., Passarella B., Guerra V., Zoccolella S., Nozzoli C., Mundi C., Leone M., Zarrelli M., Tamma F., Valluzzi F., Calabrese G., Boero G., Rini A., Traynor B. J., Singleton A. B., Mitne Neto M., Cauchi R. J., Ophoff R. A., Wiedau-Pazos M., Lomen-Hoerth C., van Deerlin V. M., Grosskreutz J., Roediger A., Gaur N., Jörk A., Barthel T., Theele E., Ilse B., Stubendorff B., Witte O. W., Steinbach R., Hübner C. A., Graff C., Brylev L., Fominykh V., Demeshonok V., Ataulina A., Rogelj B., Koritnik B., Zidar J., Ravnik-Glavač M., Glavač D., Stević Z., Drory V., Povedano M., Blair I. P., Kiernan M. C., Benyamin B., Henderson R. D., Furlong S., Mathers S., McCombe P. A., Needham M., Ngo S. T., Nicholson G. A., Pamphlett R., Rowe D. B., Steyn F. J., Williams K. L., Mather K. A., Sachdev P. S., Henders A. K., Wallace L., de Carvalho M., Pinto S., Petri S., Weber M., Rouleau G. A., Silani V., Curtis C. J., Breen G., Glass J. D., Brown R. H., Landers J. E., Shaw C. E., Andersen P. M., Groen E. J. N., van Es M. A., Pasterkamp R. J., Fan D., Garton F. C., McRae A. F., Davey Smith G., Gaunt T. R., Eberle M. A., Mill J., McLaughlin R. L., Hardiman O., Kenna K. P., Wray N. R., Tsai E., Runz H., Franke L., Al-Chalabi A., Van Damme P., van den Berg L. H., Veldink J. H., Consortium S., Consortium P., Consortium S., Consortium S., Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nature Genetics 53, 1636–1648 (2021). - PMC - PubMed
-
- DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G. Y., Karydas A., Seeley W. W., Josephs K. A., Coppola G., Geschwind D. H., Wszolek Z. K., Feldman H., Knopman D. S., Petersen R. C., Miller B. L., Dickson D. W., Boylan K. B., Graff-Radford N. R., Rademakers R., Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous