

Rational Protein Engineering to Enhance MHC-Independent T-cell Receptors
- PMID: 38980802
- PMCID: PMC11530325
- DOI: 10.1158/2159-8290.CD-23-1393
Rational Protein Engineering to Enhance MHC-Independent T-cell Receptors
Abstract
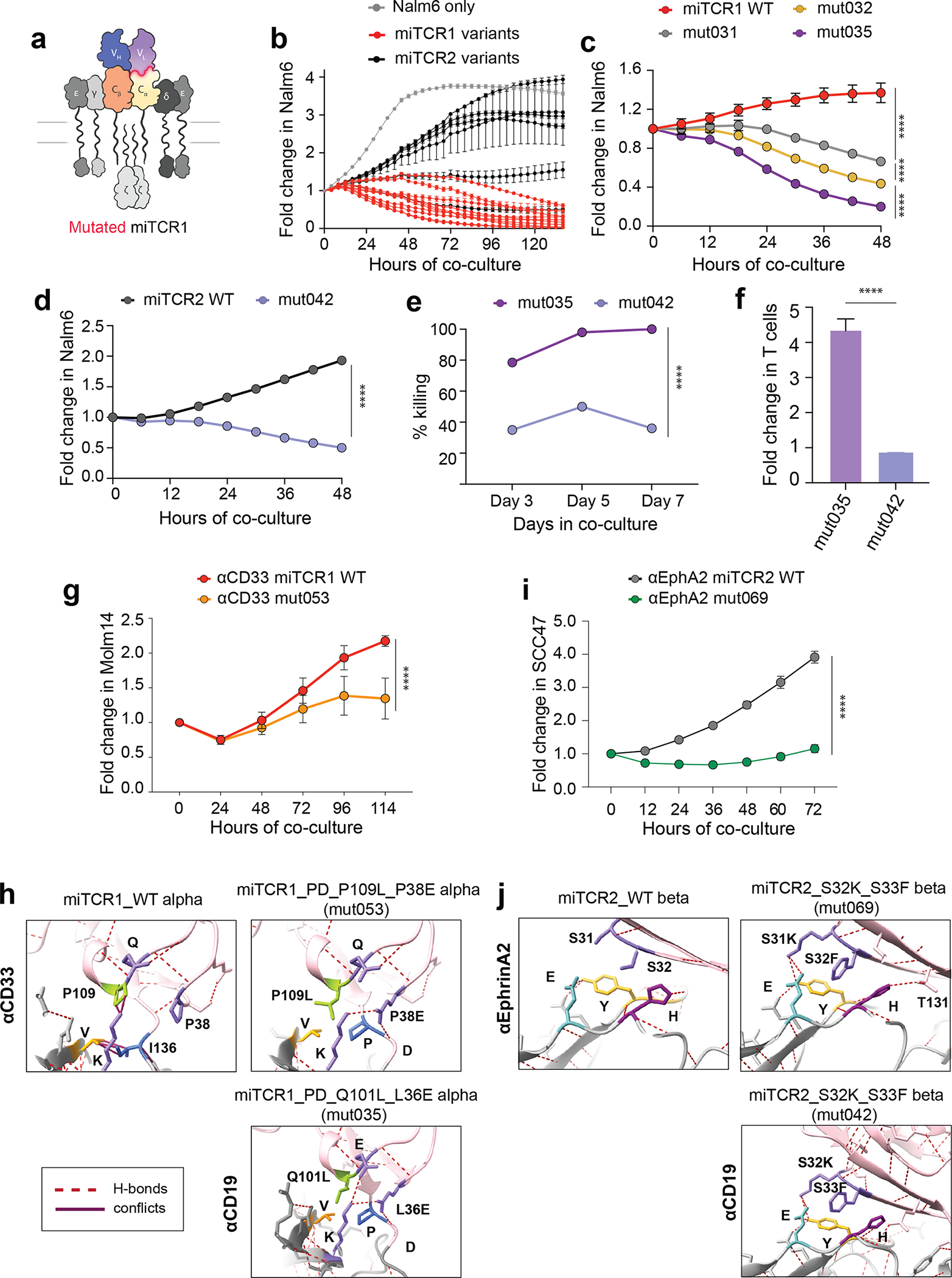
Chimeric antigen receptor (CAR)-based therapies have pioneered synthetic cellular immunity but remain limited in their long-term efficacy. Emerging data suggest that dysregulated CAR-driven T-cell activation causes T-cell dysfunction and therapeutic failure. To re-engage the precision of the endogenous T-cell response, we designed MHC-independent T-cell receptors (miTCR) by linking antibody variable domains to T-cell receptor constant chains. Using predictive modeling, we observed that this standard "cut and paste" approach to synthetic protein design resulted in myriad biochemical conflicts at the hybrid variable-constant domain interface. Through iterative modeling and sequence modifications, we developed structure-enhanced miTCRs which significantly improved receptor-driven T-cell function across multiple tumor models. We found that 41BB costimulation specifically prolonged miTCR T-cell persistence and enabled improved leukemic control in vivo compared with classic CAR T cells. Collectively, we have identified core features of hybrid receptor structure responsible for regulating function. Significance: Improving the durability of engineered T-cell immunotherapies is critical to enhancing efficacy. We used a structure-informed design to evolve improved miTCR function across several models. This work underscores the central role of synthetic receptor structure in T-cell function and provides a framework for improved receptor engineering.
©2024 American Association for Cancer Research.
Conflict of interest statement
Figures



References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
