

Thrombospondin-1 Drives Cardiac Remodeling in Chronic Kidney Disease
- PMID: 38984053
- PMCID: PMC11228122
- DOI: 10.1016/j.jacbts.2024.01.010
Thrombospondin-1 Drives Cardiac Remodeling in Chronic Kidney Disease
Abstract
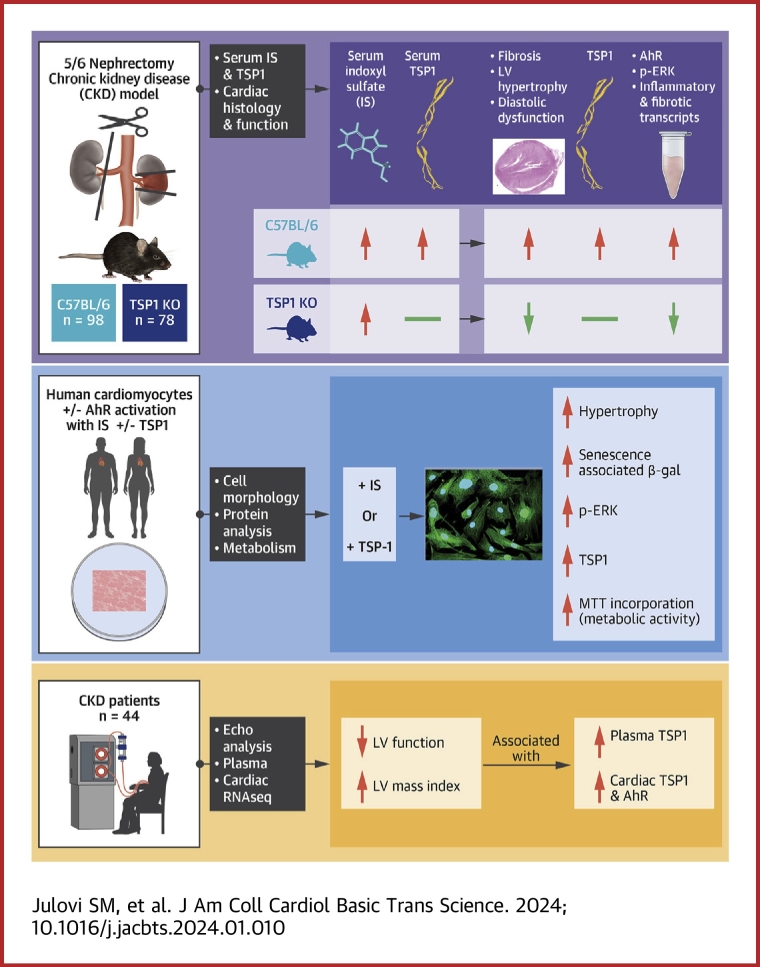
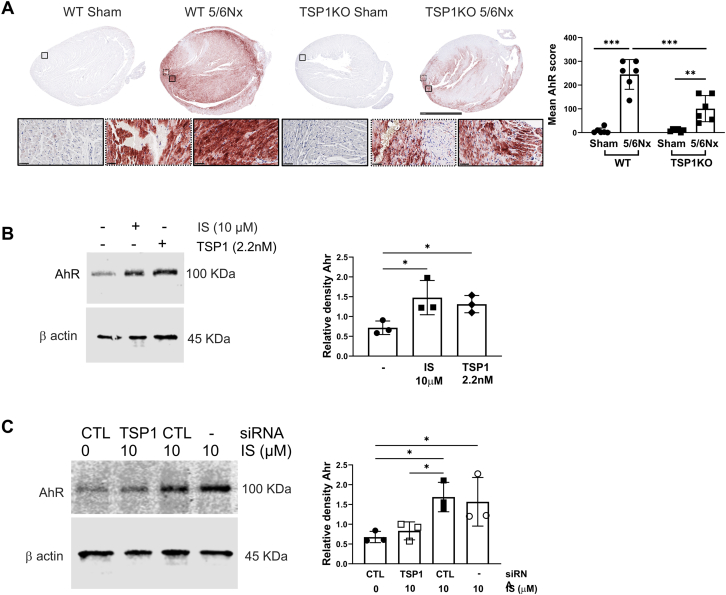
Patients with chronic kidney disease (CKD) face a high risk of cardiovascular disease. Previous studies reported that endogenous thrombospondin 1 (TSP1) involves right ventricular remodeling and dysfunction. Here we show that a murine model of CKD increased myocardial TSP1 expression and produced left ventricular hypertrophy, fibrosis, and dysfunction. TSP1 knockout mice were protected from these features. In vitro, indoxyl sulfate is driving deleterious changes in cardiomyocyte through the TSP1. In patients with CKD, TSP1 and aryl hydrocarbon receptor were both differentially expressed in the myocardium. Our findings summon large clinical studies to confirm the translational role of TSP1 in patients with CKD.
Keywords: aryl hydrocarbon receptor; cardiac fibrosis; chronic kidney disease; left ventricular hypertrophy; thrombospondin 1.
© 2024 The Authors.
Conflict of interest statement
This work was supported by a National Health Medical Research Council grant (GNT2007991) to awarded to Dr Rogers, a National Heart Foundation Vanguard Grant (106035) to Drs Rogers and Julovi, and a Westmead Medical Research Foundation grant to Dr Julovi. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.PerspectivesCOMPETENCY IN MEDICAL KNOWLEDGE: Patients with CKD continue to experience dramatically reduced life expectancy owing to concomitant CVD, termed CRS. Despite a consensus definition, there has been minimal progress in our understanding of the pathophysiology and no specific therapeutic interventions that improve outcomes. Here we demonstrate that the uremic toxin IS is driving deleterious changes in cardiomyocyte phenotype through the matrix protein TSP1. These findings provide a potential therapeutic target for CRS. TRANSLATIONAL OUTLOOK: Patients with HFpEF are vulnerable to the development of renal dysfunction during treatment for decompensation, and renal-associated mortality is higher in patients with HFpEF. We have identified a novel pathway that drives uremic toxin-induced changes in cardiac pathology through the protein TSP1. Our mechanistic data lend support for testing monoclonal antibodies or peptidic inhibitors that block the actions of TSP1 (and therefore IS), limit development of LVH, and potentially improve survival in CRS.
Figures







References
-
- Jha V., Garcia-Garcia G., Iseki K., et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382(9888):260–272. - PubMed
-
- Vanholder R., Massy Z., Argiles A., et al. Chronic kidney disease as cause of cardiovascular morbidity and mortality. Nephrol Dial Transplant. 2005;20(6):1048–1056. - PubMed
-
- Rangaswami J., Bhalla V., Blair J.E.A., et al. Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation. 2019;139(16):e840–e878. - PubMed
-
- Taveira Gomes T., Santos Araujo C., Valente F., et al. Cardiorenal syndrome and death risk in patients with heart failure or chronic kidney disease: an unmet cardiorenal need? Eur Heart J. 2021;42(suppl 1):819.
-
- Deo R., Fyr C.L., Fried L.F., et al. Health ABC study Kidney dysfunction and fatal cardiovascular disease—an association independent of atherosclerotic events: results from the Health, Aging, and Body Composition (Health ABC) study. Am Heart J. 2008;155(1):62–68. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
