

Identification and Properties of TRPV4 Mutant Channels Present in Polycystic Kidney Disease Patients
- PMID: 38984987
- PMCID: PMC11384909
- DOI: 10.1093/function/zqae031
Identification and Properties of TRPV4 Mutant Channels Present in Polycystic Kidney Disease Patients
Abstract
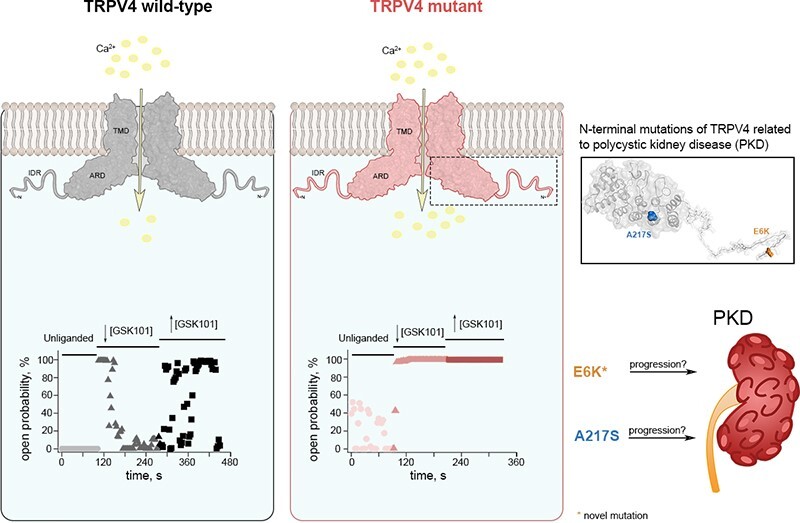
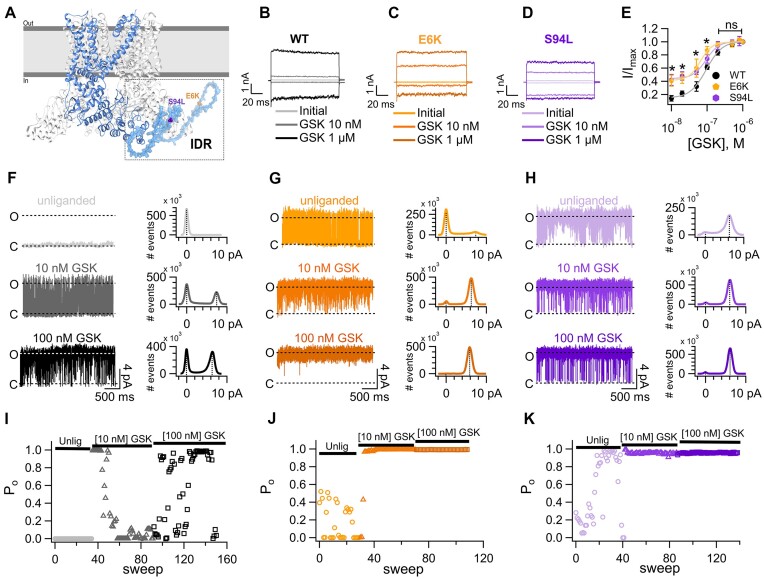
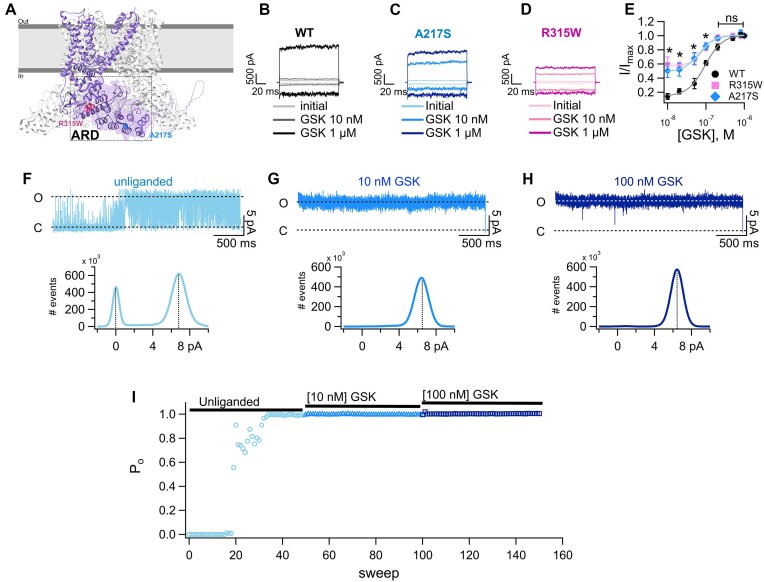
Polycystic kidney disease (PKD), a disease characterized by the enlargement of the kidney through cystic growth is the fourth leading cause of end-stage kidney disease world-wide. Transient receptor potential Vanilloid 4 (TRPV4), a calcium-permeable TRP, channel participates in kidney cell physiology and since TRPV4 forms complexes with another channel whose malfunction is associated to PKD, TRPP2 (or PKD2), we sought to determine whether patients with PKD, exhibit previously unknown mutations in TRPV4. Here, we report the presence of mutations in the TRPV4 gene in patients diagnosed with PKD and determine that they produce gain-of-function (GOF). Mutations in the sequence of the TRPV4 gene have been associated to a broad spectrum of neuropathies and skeletal dysplasias but not PKD, and their biophysical effects on channel function have not been elucidated. We identified and examined the functional behavior of a novel E6K mutant and of the previously known S94L and A217S mutant TRVP4 channels. The A217S mutation has been associated to mixed neuropathy and/or skeletal dysplasia phenotypes, however, the PKD carriers of these variants had not been diagnosed with these reported clinical manifestations. The presence of certain mutations in TRPV4 may influence the progression and severity of PKD through GOF mechanisms. PKD patients carrying TRVP4 mutations are putatively more likely to require dialysis or renal transplant as compared to those without these mutations.
Keywords: PKD; TRPV4; channelopathies; gain-of-function; ion channels; kidney disease; polycystic kidney disease; renal function.
© The Author(s) 2024. Published by Oxford University Press on behalf of American Physiological Society.
Conflict of interest statement
None declared.
Figures
Similar articles
-
Loss of primary cilia increases polycystin-2 and TRPV4 and the appearance of a nonselective cation channel in the mouse cortical collecting duct.Am J Physiol Renal Physiol. 2019 Sep 1;317(3):F632-F637. doi: 10.1152/ajprenal.00210.2019. Epub 2019 Jul 17. Am J Physiol Renal Physiol. 2019. PMID: 31313950 Free PMC article.
-
TRPV4 mutations causing mixed neuropathy and skeletal phenotypes result in severe gain of function.Ann Clin Transl Neurol. 2022 Mar;9(3):375-391. doi: 10.1002/acn3.51523. Epub 2022 Feb 16. Ann Clin Transl Neurol. 2022. PMID: 35170874 Free PMC article.
-
Molecular analysis of a consanguineous Iranian polycystic kidney disease family identifies a PKD2 mutation that aids diagnostics.BMC Nephrol. 2013 Sep 8;14:190. doi: 10.1186/1471-2369-14-190. BMC Nephrol. 2013. PMID: 24011172 Free PMC article.
-
Ciliary Ion Channels in Polycystic Kidney Disease.Cells. 2025 Mar 19;14(6):459. doi: 10.3390/cells14060459. Cells. 2025. PMID: 40136708 Free PMC article. Review.
-
Roles for TRPV4 in disease: A discussion of possible mechanisms.Cell Calcium. 2024 Dec;124:102972. doi: 10.1016/j.ceca.2024.102972. Epub 2024 Nov 22. Cell Calcium. 2024. PMID: 39609180 Review.
References
-
- Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet North Am Ed. 2019;393(10174):919–935. - PubMed
-
- Mantovani V, Bin S, Graziano C et al. Gene panel analysis in a large cohort of patients with autosomal dominant polycystic kidney disease allows the identification of 80 potentially causative novel variants and the characterization of a complex genetic architecture in a subset of families. Front Genet. 2020;11:464. - PMC - PubMed
-
- Nauli SM, Alenghat FJ, Luo Y et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous