

Co-analysis of methylation platforms for signatures of biological aging in the domestic dog reveals previously unexplored confounding factors
- PMID: 38985449
- PMCID: PMC11272130
- DOI: 10.18632/aging.206012
Co-analysis of methylation platforms for signatures of biological aging in the domestic dog reveals previously unexplored confounding factors
Abstract
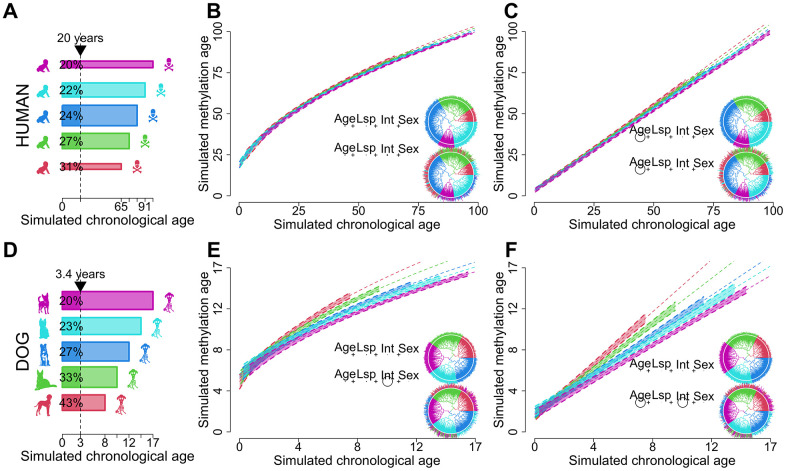
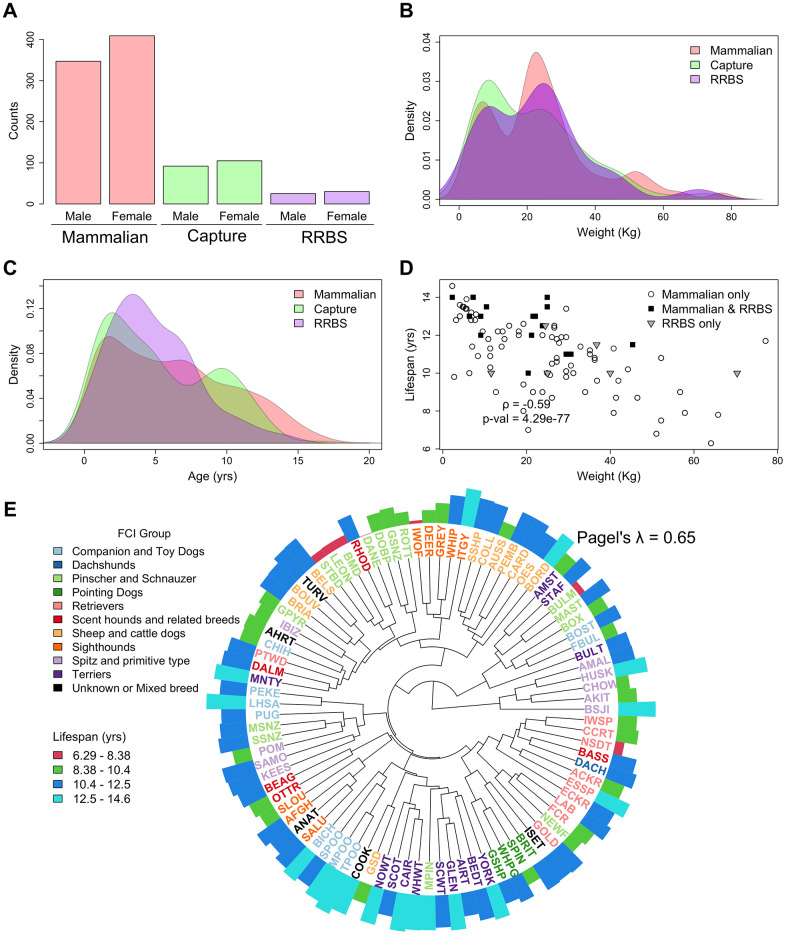
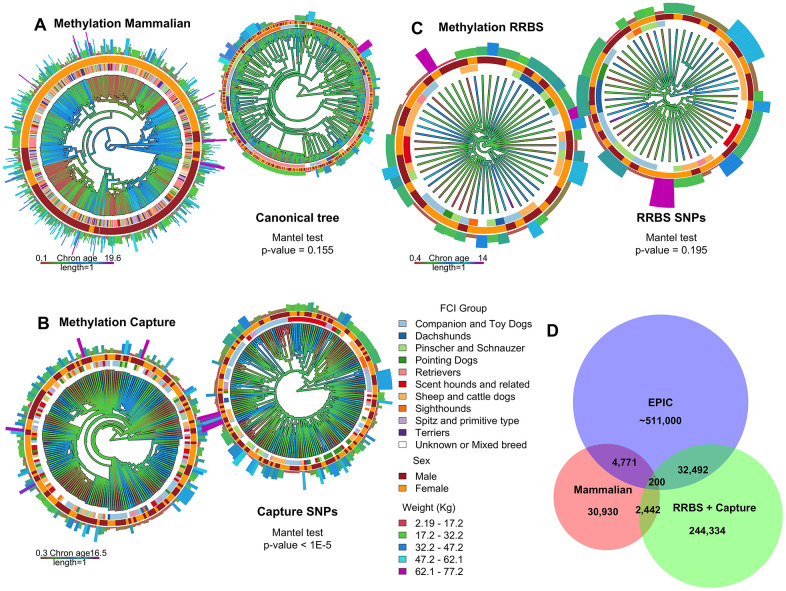
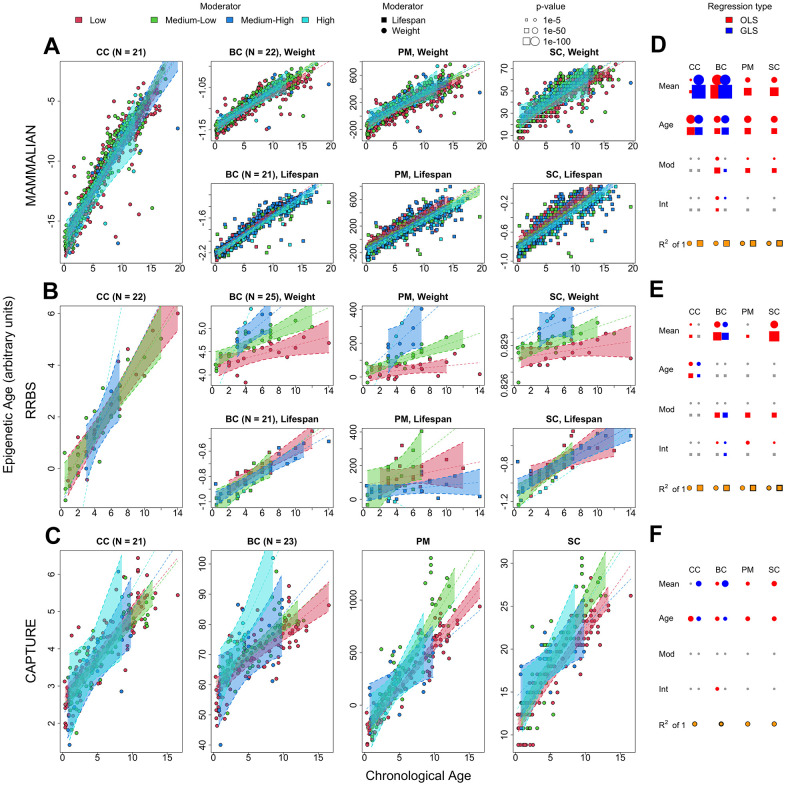
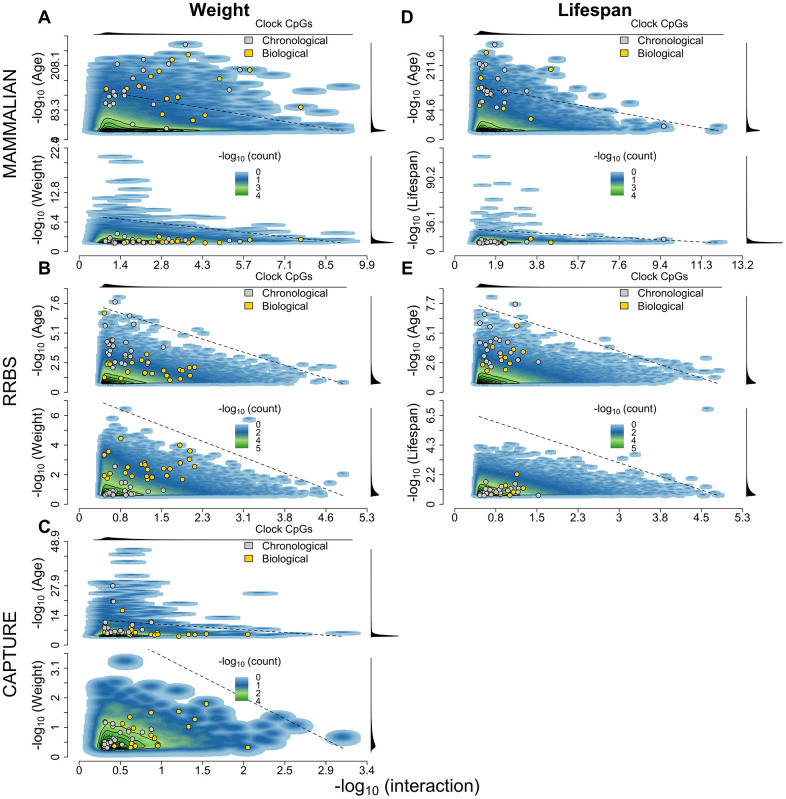
Chronological age reveals the number of years an individual has lived since birth. By contrast, biological age varies between individuals of the same chronological age at a rate reflective of physiological decline. Differing rates of physiological decline are related to longevity and result from genetics, environment, behavior, and disease. The creation of methylation biological age predictors is a long-standing challenge in aging research due to the lack of individual pre-mortem longevity data. The consistent differences in longevity between domestic dog breeds enable the construction of biological age estimators which can, in turn, be contrasted with methylation measurements to elucidate mechanisms of biological aging. We draw on three flagship methylation studies using distinct measurement platforms and tissues to assess the feasibility of creating biological age methylation clocks in the dog. We expand epigenetic clock building strategies to accommodate phylogenetic relationships between individuals, thus controlling for the use of breed standard metrics. We observe that biological age methylation clocks are affected by population stratification and require heavy parameterization to achieve effective predictions. Finally, we observe that methylation-related markers reflecting biological age signals are rare and do not colocalize between datasets.
Keywords: biological age; dog; lifespan; methylation; penalized regression.
Conflict of interest statement
Figures
Similar articles
-
DNA methylation clocks for dogs and humans.Proc Natl Acad Sci U S A. 2022 May 24;119(21):e2120887119. doi: 10.1073/pnas.2120887119. Epub 2022 May 17. Proc Natl Acad Sci U S A. 2022. PMID: 35580182 Free PMC article.
-
DNA Methylation Clocks in Aging: Categories, Causes, and Consequences.Mol Cell. 2018 Sep 20;71(6):882-895. doi: 10.1016/j.molcel.2018.08.008. Mol Cell. 2018. PMID: 30241605 Free PMC article. Review.
-
An epigenetic aging clock for dogs and wolves.Aging (Albany NY). 2017 Mar 28;9(3):1055-1068. doi: 10.18632/aging.101211. Aging (Albany NY). 2017. PMID: 28373601 Free PMC article.
-
Associations of four biological age markers with child development: A multi-omic analysis in the European HELIX cohort.Elife. 2023 Jun 6;12:e85104. doi: 10.7554/eLife.85104. Elife. 2023. PMID: 37278618 Free PMC article.
-
Individual DNA Methylation Profile is Correlated with Age and can be Targeted to Modulate Healthy Aging and Longevity.Curr Pharm Des. 2019;25(39):4139-4149. doi: 10.2174/1381612825666191112095655. Curr Pharm Des. 2019. PMID: 31721695 Review.
References
-
- Horvath S, Haghani A, Macoretta N, Ablaeva J, Zoller JA, Li CZ, Zhang J, Takasugi M, Zhao Y, Rydkina E, Zhang Z, Emmrich S, Raj K, et al.. DNA methylation clocks tick in naked mole rats but queens age more slowly than nonbreeders. Nat Aging. 2022; 2:46–59. 10.1038/s43587-021-00152-1 - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
