

Repeated plague infections across six generations of Neolithic Farmers
- PMID: 38987589
- PMCID: PMC11291285
- DOI: 10.1038/s41586-024-07651-2
Repeated plague infections across six generations of Neolithic Farmers
Abstract
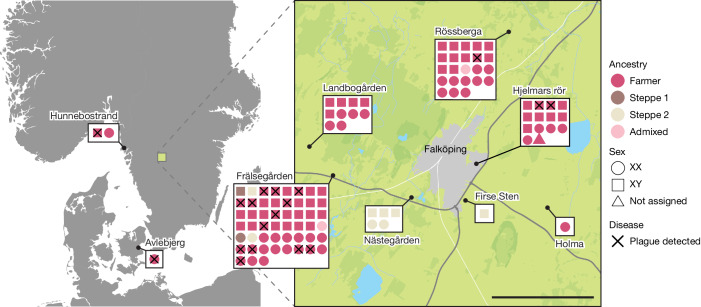
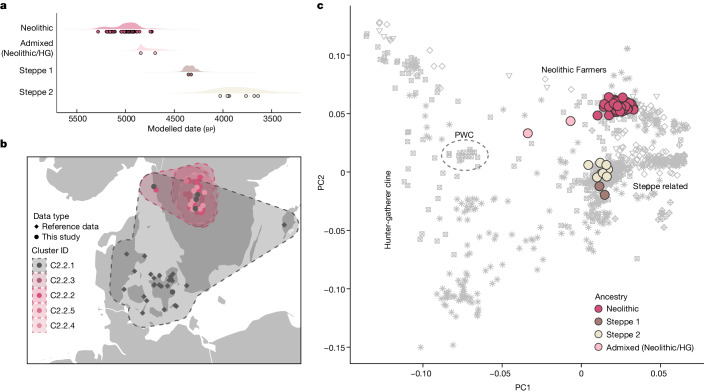
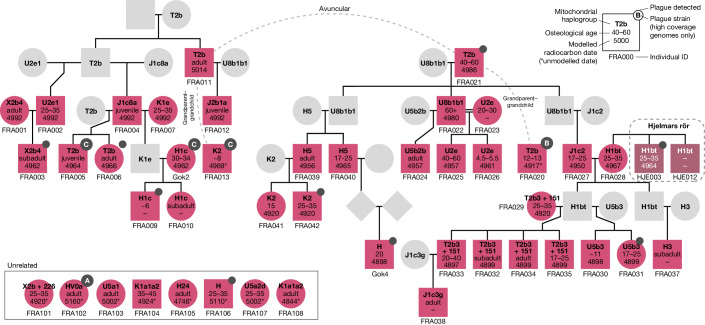
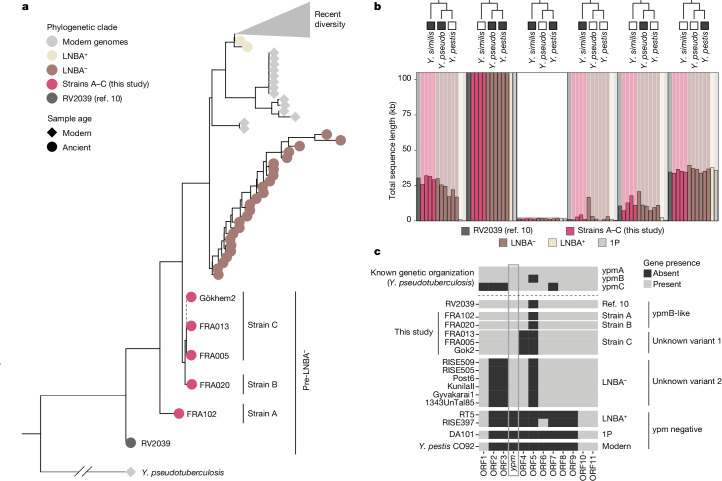
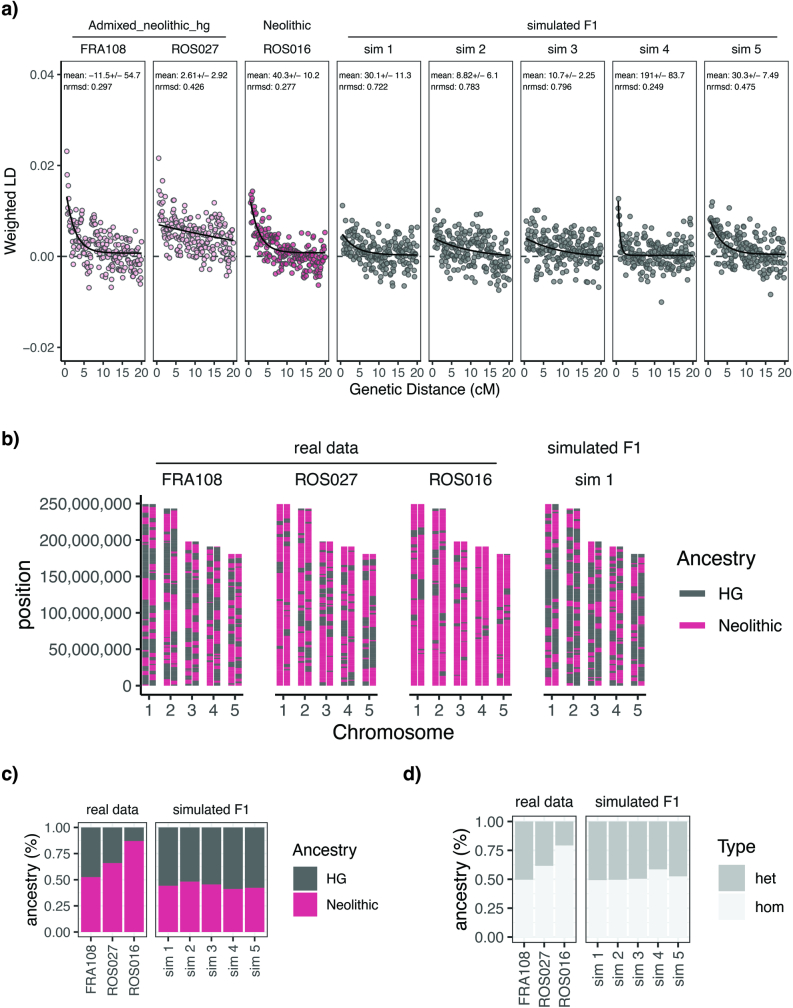
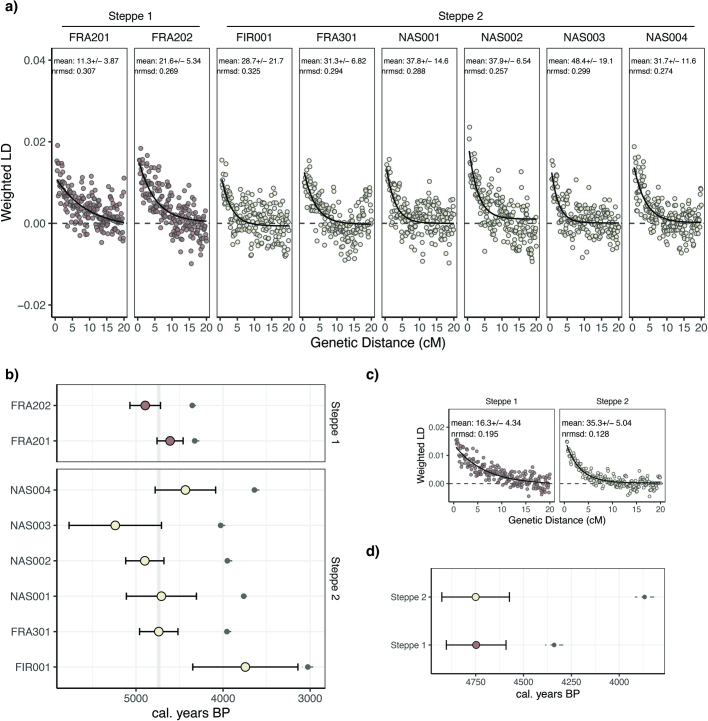
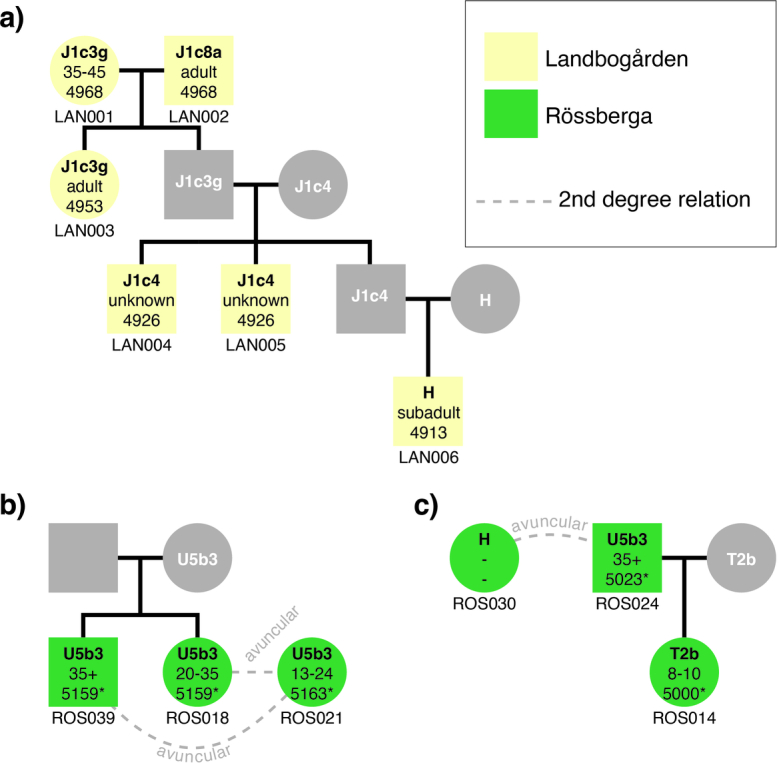
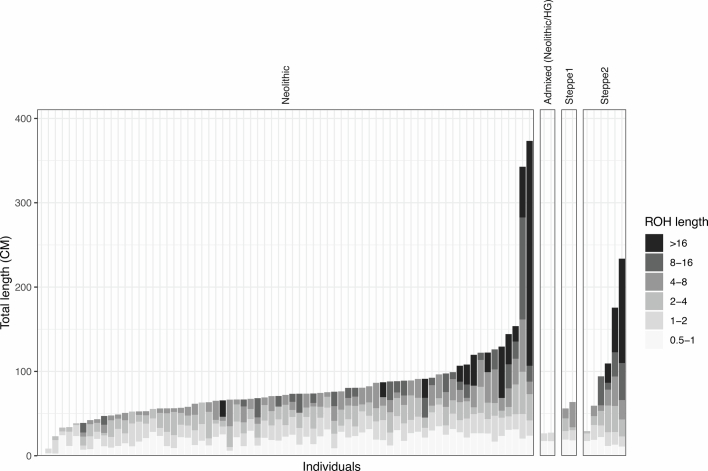
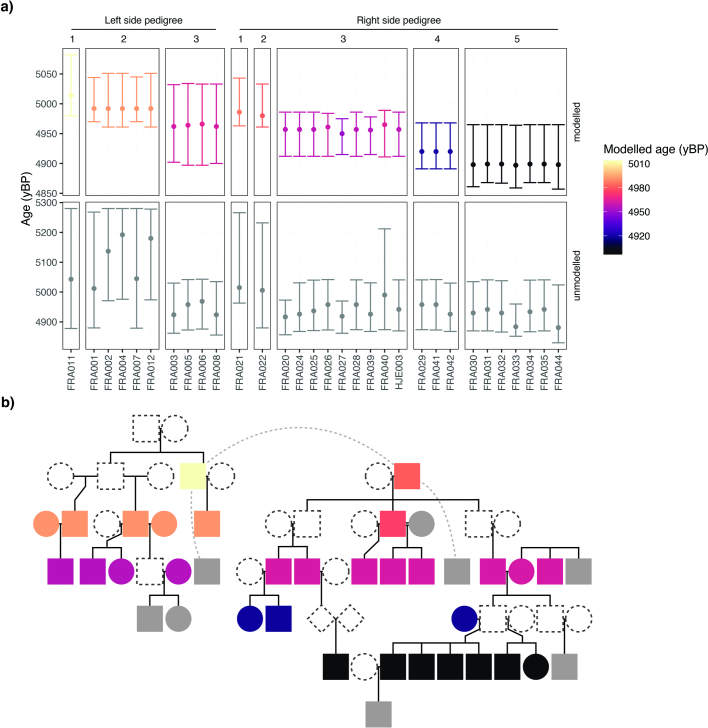
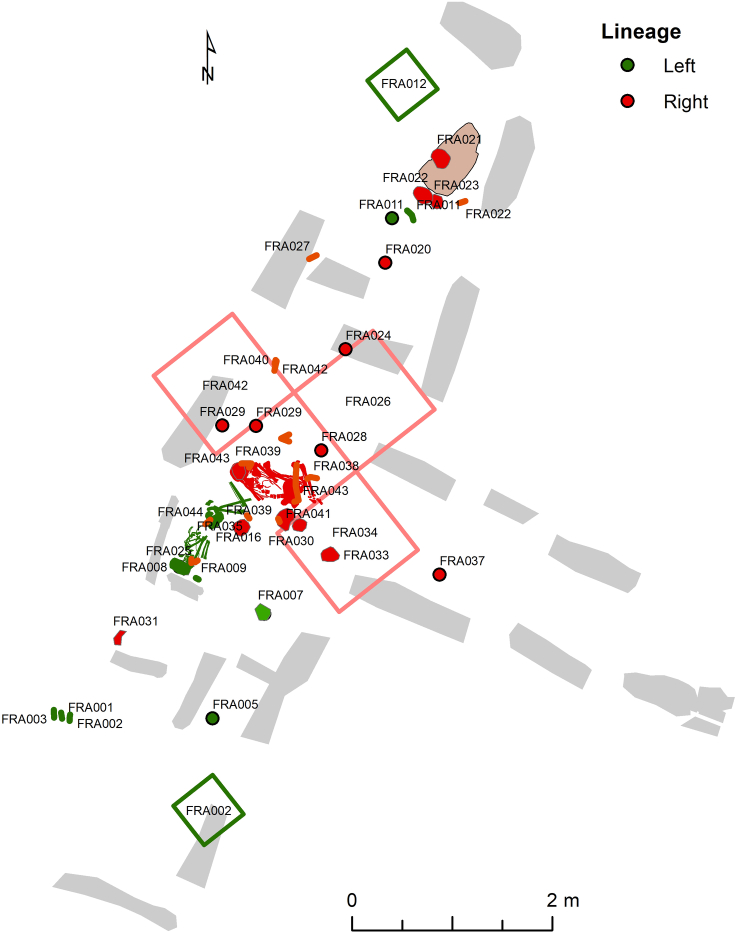
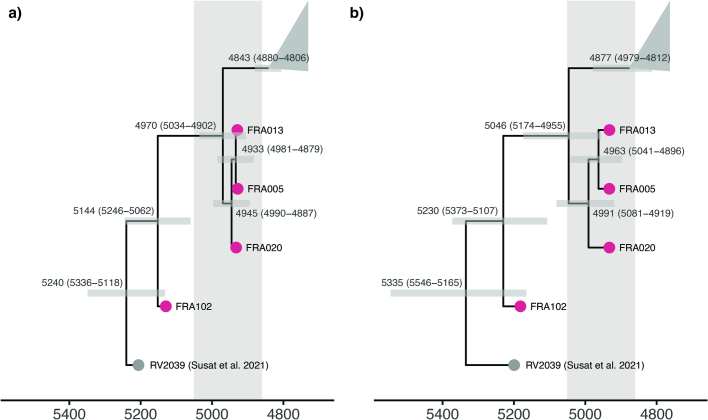
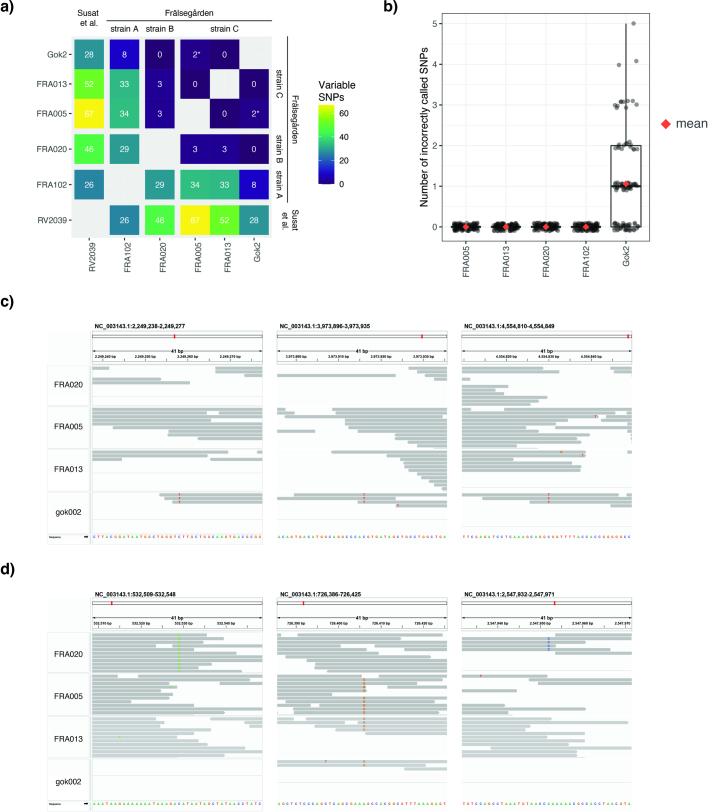
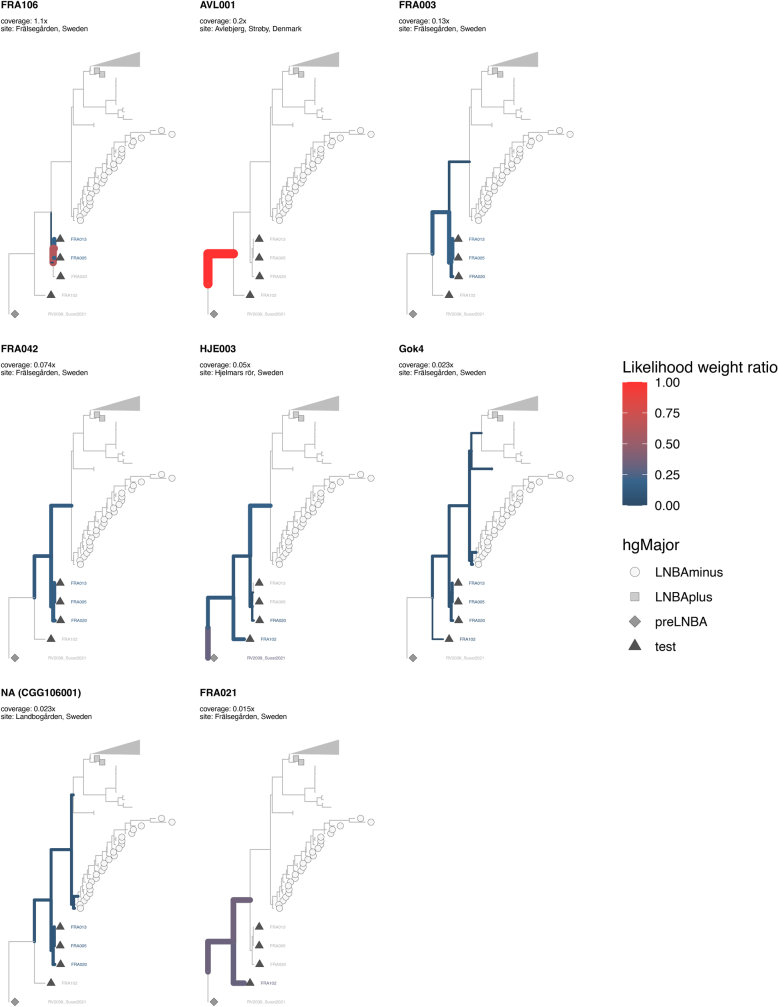
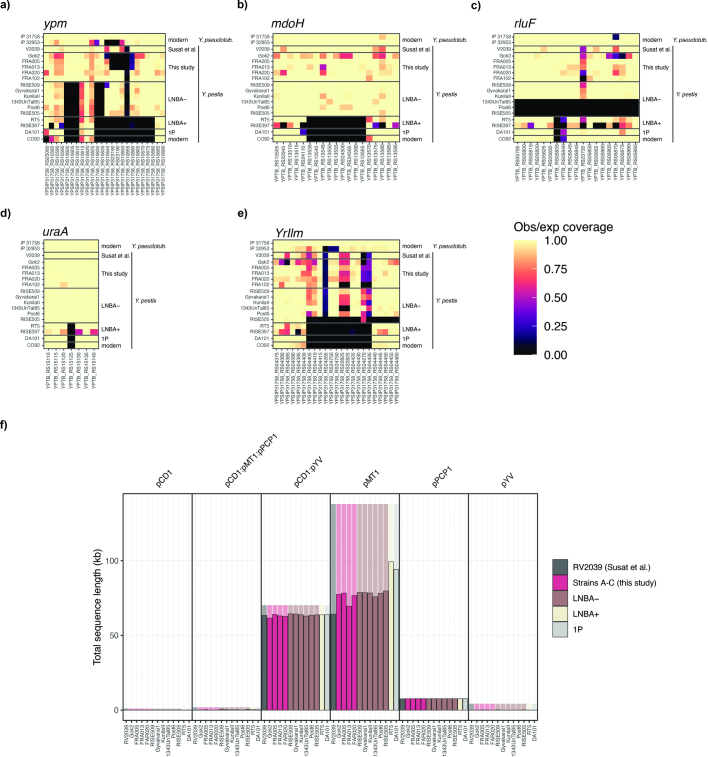
In the period between 5,300 and 4,900 calibrated years before present (cal. BP), populations across large parts of Europe underwent a period of demographic decline1,2. However, the cause of this so-called Neolithic decline is still debated. Some argue for an agricultural crisis resulting in the decline3, others for the spread of an early form of plague4. Here we use population-scale ancient genomics to infer ancestry, social structure and pathogen infection in 108 Scandinavian Neolithic individuals from eight megalithic graves and a stone cist. We find that the Neolithic plague was widespread, detected in at least 17% of the sampled population and across large geographical distances. We demonstrate that the disease spread within the Neolithic community in three distinct infection events within a period of around 120 years. Variant graph-based pan-genomics shows that the Neolithic plague genomes retained ancestral genomic variation present in Yersinia pseudotuberculosis, including virulence factors associated with disease outcomes. In addition, we reconstruct four multigeneration pedigrees, the largest of which consists of 38 individuals spanning six generations, showing a patrilineal social organization. Lastly, we document direct genomic evidence for Neolithic female exogamy in a woman buried in a different megalithic tomb than her brothers. Taken together, our findings provide a detailed reconstruction of plague spread within a large patrilineal kinship group and identify multiple plague infections in a population dated to the beginning of the Neolithic decline.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kristiansen, K. in The Oxford Handbook of Neolithic Europe (eds Fowler, C. et al.) Ch. 56 (Oxford Univ. Press, 2014).
-
- Müller, J. in Environment and subsistence - forty years after Janusz Kruk’s “Settlement studies” (eds Kadrow, S. & Włodarczak, P.) 493–506 (Dr. Rudolf Habelt GmbH, 2013).
-
- Colledge, S., Conolly, J., Crema, E. & Shennan, S. Neolithic population crash in northwest Europe associated with agricultural crisis. Quat. Res.92, 686–707 (2019).
-
- Rascovan, N. et al. Emergence and spread of basal lineages of Yersinia pestis during the Neolithic Decline. Cell176, 295–305 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
