

Glutamate acts on acid-sensing ion channels to worsen ischaemic brain injury
- PMID: 38987597
- PMCID: PMC11269185
- DOI: 10.1038/s41586-024-07684-7
Glutamate acts on acid-sensing ion channels to worsen ischaemic brain injury
Abstract
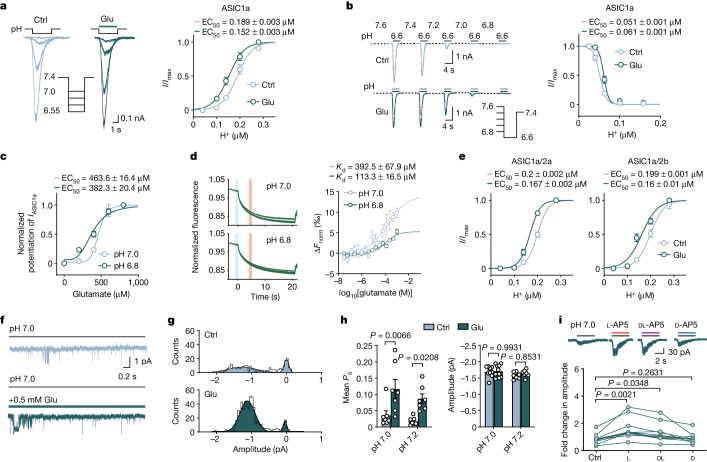
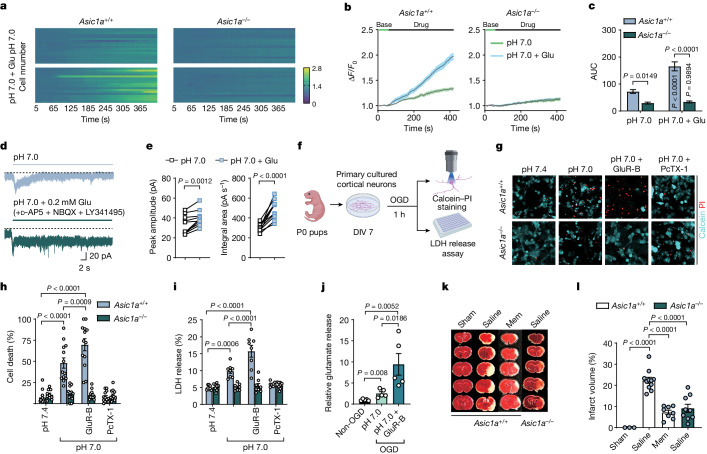
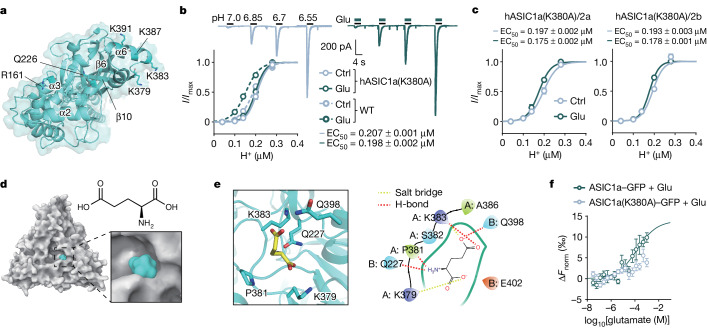
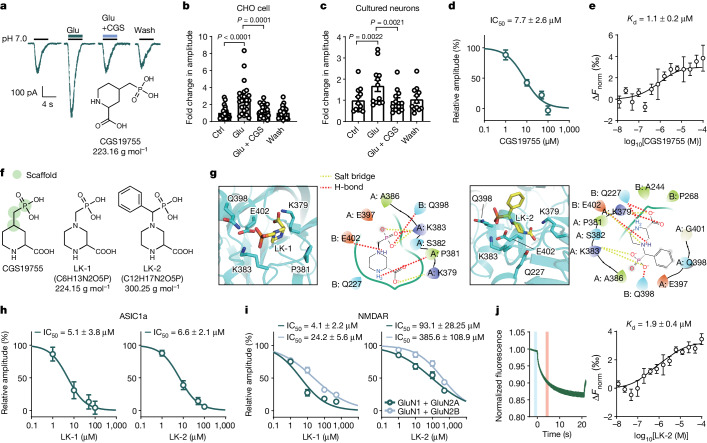
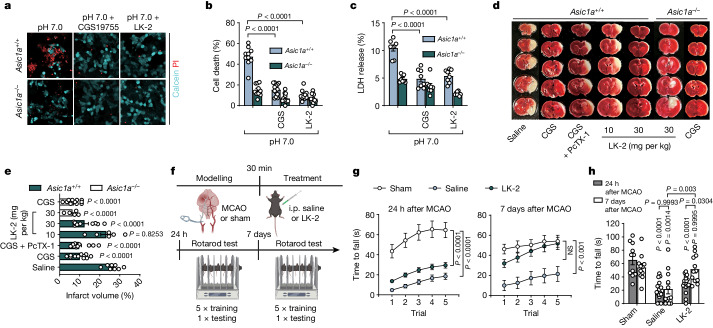
Glutamate is traditionally viewed as the first messenger to activate NMDAR (N-methyl-D-aspartate receptor)-dependent cell death pathways in stroke1,2, but unsuccessful clinical trials with NMDAR antagonists implicate the engagement of other mechanisms3-7. Here we show that glutamate and its structural analogues, including NMDAR antagonist L-AP5 (also known as APV), robustly potentiate currents mediated by acid-sensing ion channels (ASICs) associated with acidosis-induced neurotoxicity in stroke4. Glutamate increases the affinity of ASICs for protons and their open probability, aggravating ischaemic neurotoxicity in both in vitro and in vivo models. Site-directed mutagenesis, structure-based modelling and functional assays reveal a bona fide glutamate-binding cavity in the extracellular domain of ASIC1a. Computational drug screening identified a small molecule, LK-2, that binds to this cavity and abolishes glutamate-dependent potentiation of ASIC currents but spares NMDARs. LK-2 reduces the infarct volume and improves sensorimotor recovery in a mouse model of ischaemic stroke, reminiscent of that seen in mice with Asic1a knockout or knockout of other cation channels4-7. We conclude that glutamate functions as a positive allosteric modulator for ASICs to exacerbate neurotoxicity, and preferential targeting of the glutamate-binding site on ASICs over that on NMDARs may be strategized for developing stroke therapeutics lacking the psychotic side effects of NMDAR antagonists.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures










References
-
- Dirnagl, U., Iadecola, C. & Moskowitz, M. A. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci.22, 391–397 (1999). - PubMed
-
- Lee, J. M., Zipfel, G. J. & Choi, D. W. The changing landscape of ischaemic brain injury mechanisms. Nature399, A7–A14 (1999). - PubMed
-
- O’Collins, V. E. et al. 1,026 experimental treatments in acute stroke. Ann. Neurol.59, 467–477 (2006). - PubMed
-
- Xiong, Z. G. et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell118, 687–698 (2004). - PubMed
-
- Gao, J. et al. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron48, 635–646 (2005). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
