

The Open Force Field Initiative: Open Software and Open Science for Molecular Modeling
- PMID: 38989715
- PMCID: PMC11284797
- DOI: 10.1021/acs.jpcb.4c01558
The Open Force Field Initiative: Open Software and Open Science for Molecular Modeling
Abstract
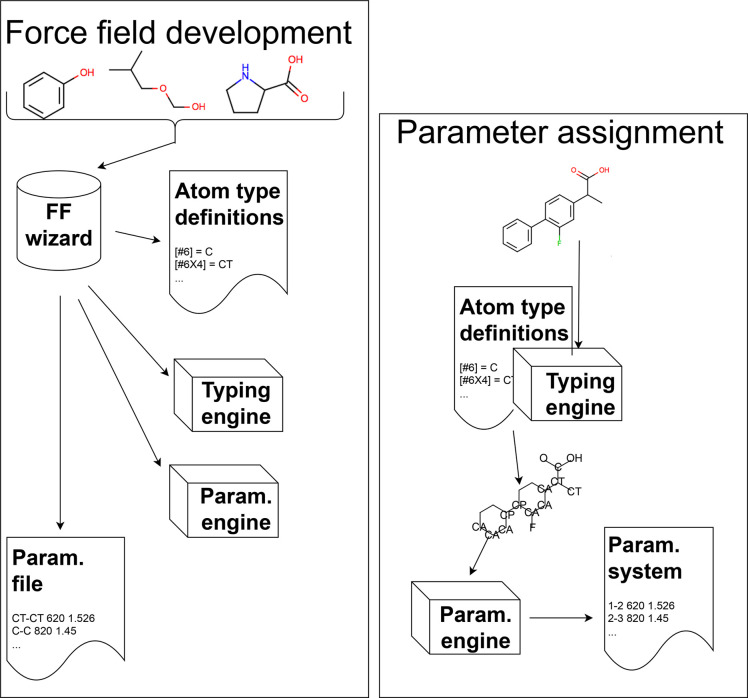
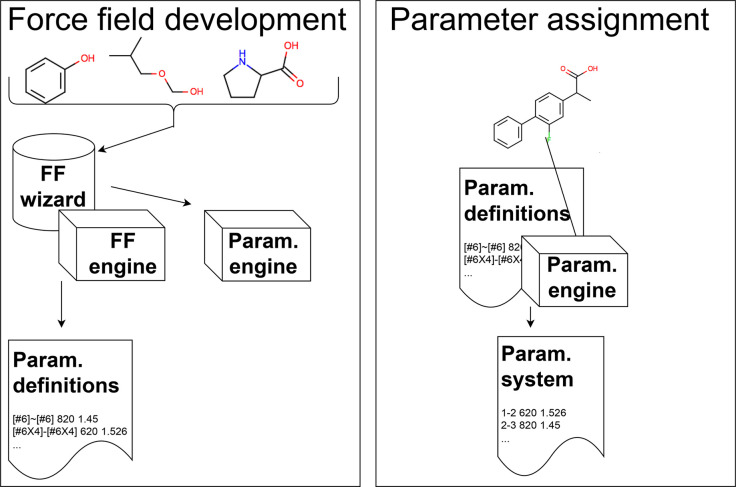
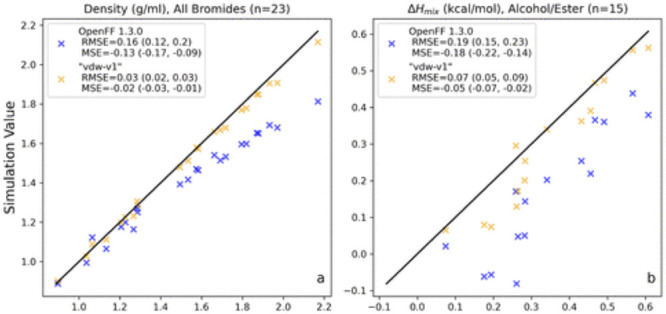
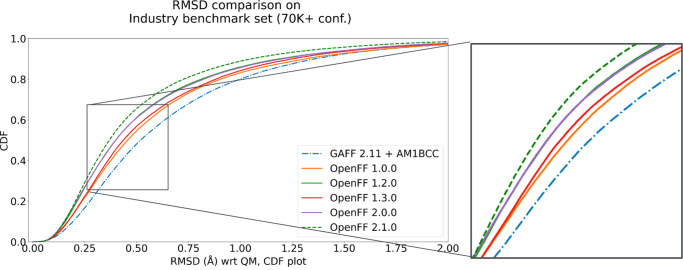
Force fields are a key component of physics-based molecular modeling, describing the energies and forces in a molecular system as a function of the positions of the atoms and molecules involved. Here, we provide a review and scientific status report on the work of the Open Force Field (OpenFF) Initiative, which focuses on the science, infrastructure and data required to build the next generation of biomolecular force fields. We introduce the OpenFF Initiative and the related OpenFF Consortium, describe its approach to force field development and software, and discuss accomplishments to date as well as future plans. OpenFF releases both software and data under open and permissive licensing agreements to enable rapid application, validation, extension, and modification of its force fields and software tools. We discuss lessons learned to date in this new approach to force field development. We also highlight ways that other force field researchers can get involved, as well as some recent successes of outside researchers taking advantage of OpenFF tools and data.
Conflict of interest statement
The authors declare the following competing financial interest(s): DLM serves on the scientific advisory boards of OpenEye Scientific Software and Anagenex, and is an Open Science Fellow with Psivant Therapeutics. MRS an Open Science Fellow with Psivant Therapeutics and consults for Relay Therapeutics. MKG has an equity interest in and is a cofounder and scientific advisor of VeraChem LLC; and is an advisor to Denovicon Therapeutics and InCerebro Inc. YW has limited financial interest in Flagship Pioneering, Inc., and its subsidiaries.
Figures
Similar articles
-
Collaborative Assessment of Molecular Geometries and Energies from the Open Force Field.J Chem Inf Model. 2022 Dec 12;62(23):6094-6104. doi: 10.1021/acs.jcim.2c01185. Epub 2022 Nov 26. J Chem Inf Model. 2022. PMID: 36433835 Free PMC article.
-
Development and Benchmarking of Open Force Field 2.0.0: The Sage Small Molecule Force Field.J Chem Theory Comput. 2023 Jun 13;19(11):3251-3275. doi: 10.1021/acs.jctc.3c00039. Epub 2023 May 11. J Chem Theory Comput. 2023. PMID: 37167319 Free PMC article.
-
Benchmark assessment of molecular geometries and energies from small molecule force fields.F1000Res. 2020 Dec 3;9:Chem Inf Sci-1390. doi: 10.12688/f1000research.27141.1. eCollection 2020. F1000Res. 2020. PMID: 33604023 Free PMC article.
-
Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?J Comput Aided Mol Des. 2019 Feb;33(2):133-203. doi: 10.1007/s10822-018-0111-4. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506158 Review.
-
Data science techniques in biomolecular force field development.Curr Opin Struct Biol. 2023 Feb;78:102502. doi: 10.1016/j.sbi.2022.102502. Epub 2022 Nov 30. Curr Opin Struct Biol. 2023. PMID: 36462448 Review.
Cited by
-
How does machine learning augment alchemical binding free energy calculations?Future Med Chem. 2025 Mar;17(5):509-511. doi: 10.1080/17568919.2025.2463870. Epub 2025 Feb 8. Future Med Chem. 2025. PMID: 39922803 No abstract available.
-
Advances and Challenges in Milestoning Simulations for Drug-Target Kinetics.J Chem Theory Comput. 2024 Nov 26;20(22):9759-9769. doi: 10.1021/acs.jctc.4c01108. Epub 2024 Nov 7. J Chem Theory Comput. 2024. PMID: 39508322 Free PMC article.
-
Fine-tuning molecular mechanics force fields to experimental free energy measurements.bioRxiv [Preprint]. 2025 Jan 8:2025.01.06.631610. doi: 10.1101/2025.01.06.631610. bioRxiv. 2025. PMID: 39829785 Free PMC article. Preprint.
-
Data-driven parametrization of molecular mechanics force fields for expansive chemical space coverage.Chem Sci. 2024 Dec 31;16(6):2730-2740. doi: 10.1039/d4sc06640e. eCollection 2025 Feb 5. Chem Sci. 2024. PMID: 39802691 Free PMC article.
-
Grappa - a machine learned molecular mechanics force field.Chem Sci. 2025 Jan 15;16(6):2907-2930. doi: 10.1039/d4sc05465b. eCollection 2025 Feb 5. Chem Sci. 2025. PMID: 39822899 Free PMC article.
References
-
- Ponder J. W.; Case D. A.. Advances in Protein Chemistry; Protein Simulations; Academic Press, 2003; Vol. 66; pp 27–85. - PubMed
-
- Chipot C., Pohorille A., Eds. Free Energy Calculations: Theory and Applications in Chemistry and Biology; Springer Series in Chemical Physics; Springer-Verlag: Berlin Heidelberg, 2007.
