

Full-genome sequencing of dozens of new DNA viruses found in Spanish bat feces
- PMID: 38990026
- PMCID: PMC11323972
- DOI: 10.1128/spectrum.00675-24
Full-genome sequencing of dozens of new DNA viruses found in Spanish bat feces
Abstract
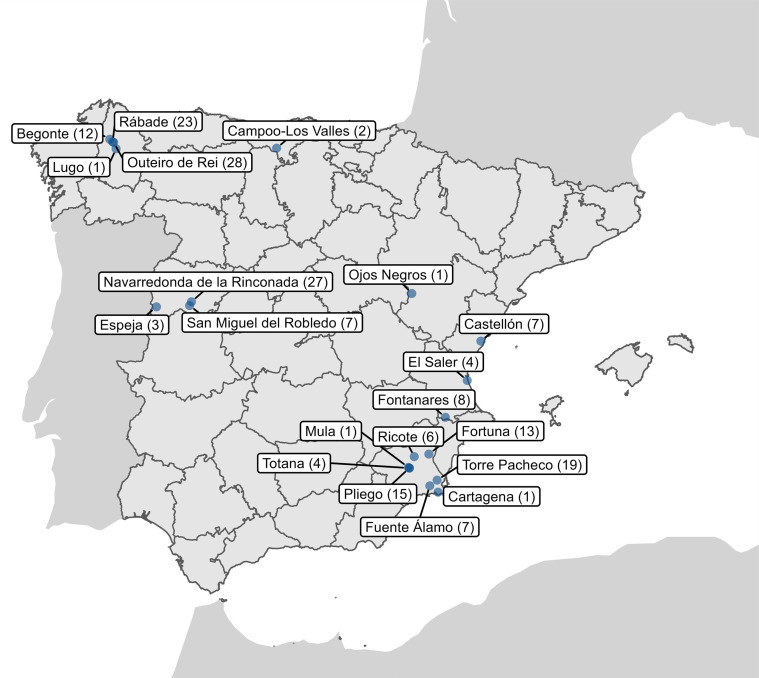
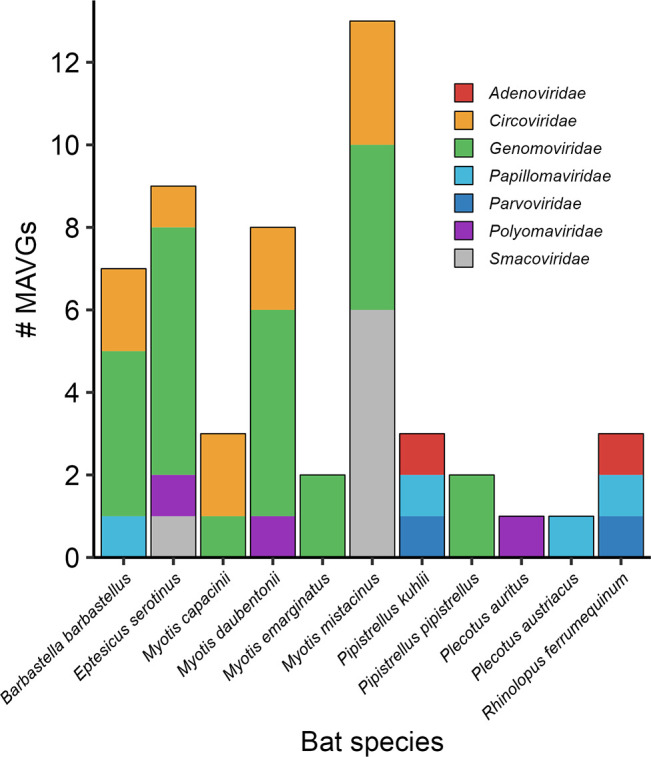
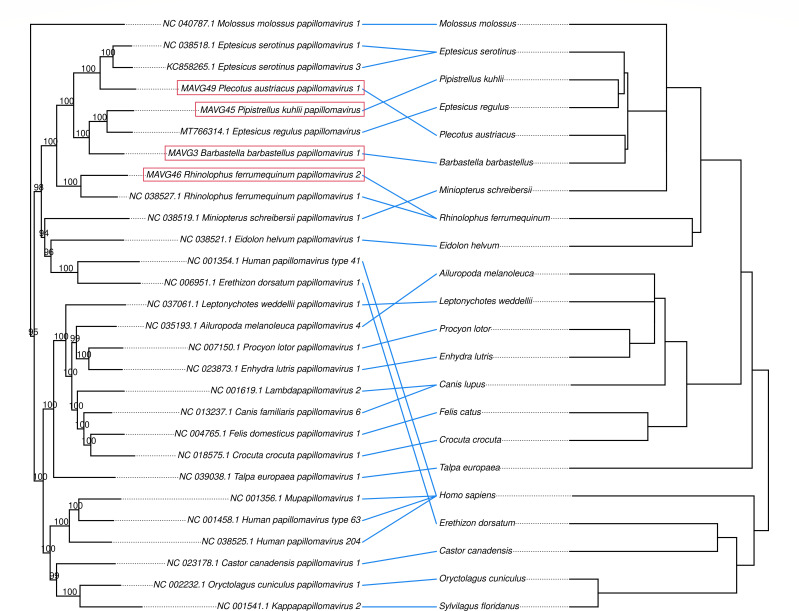
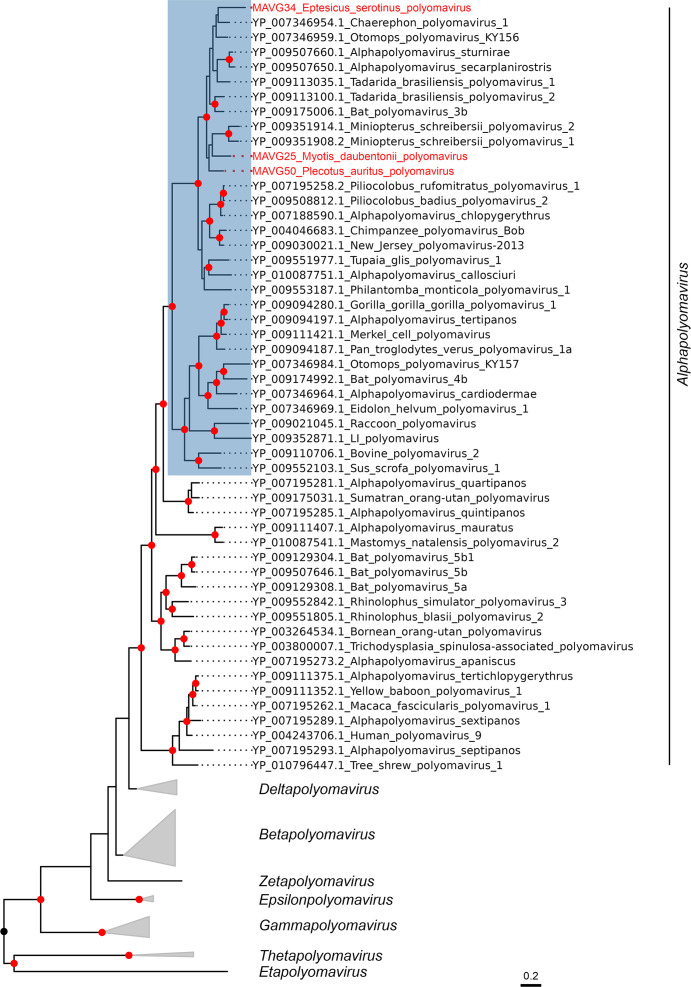
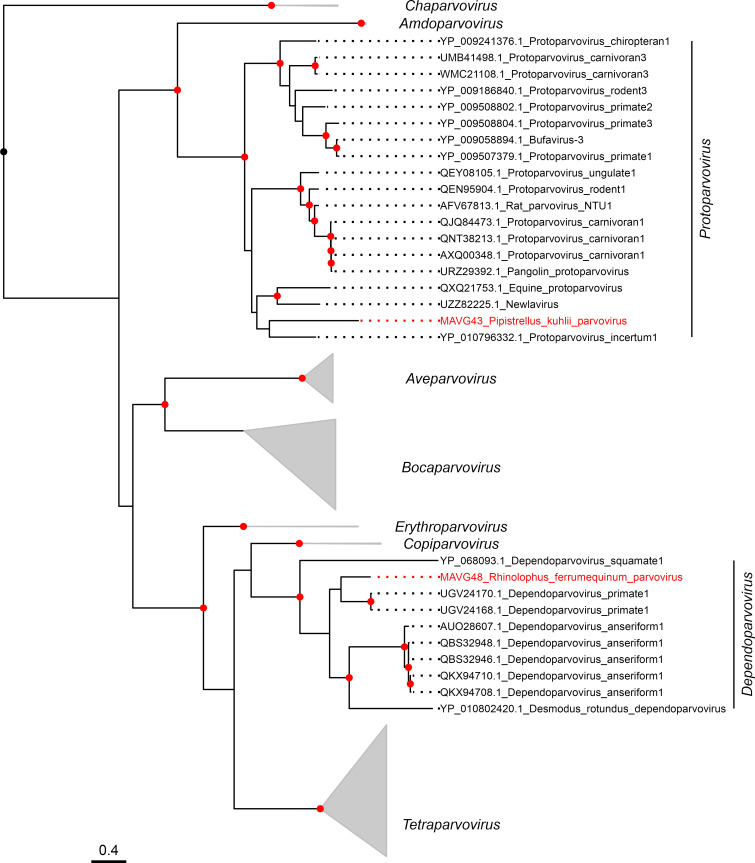
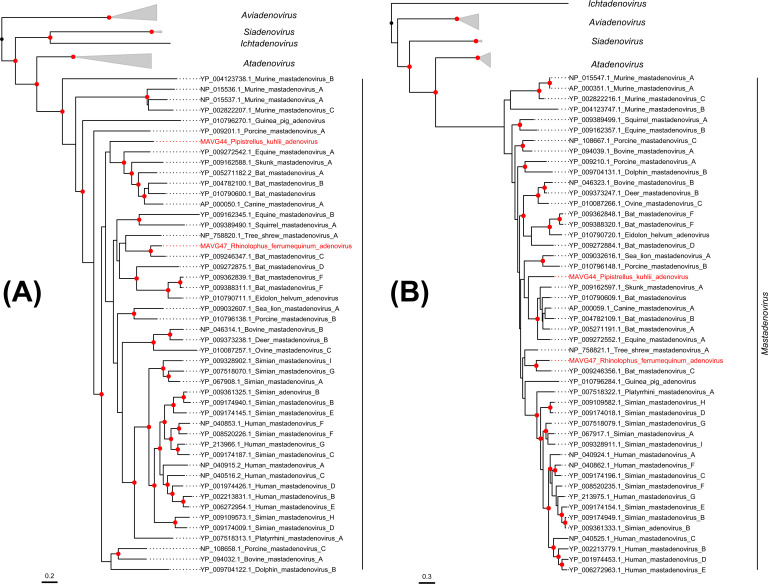
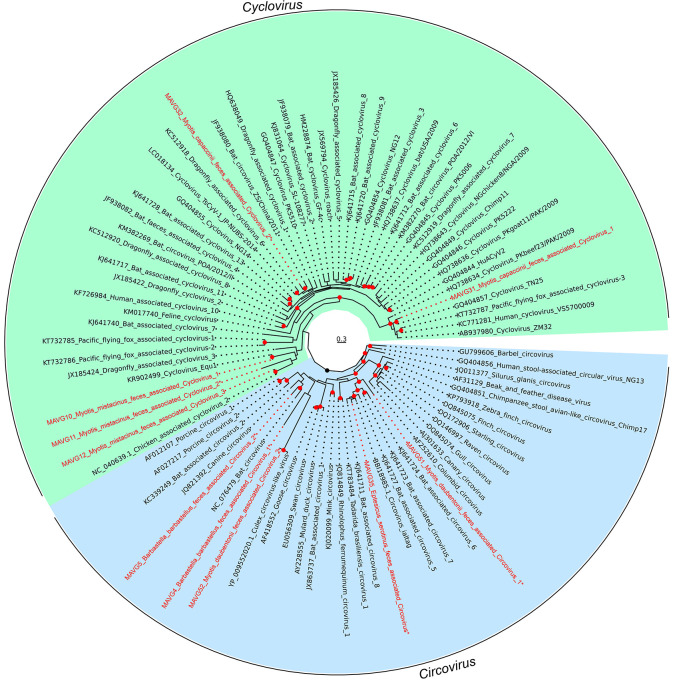
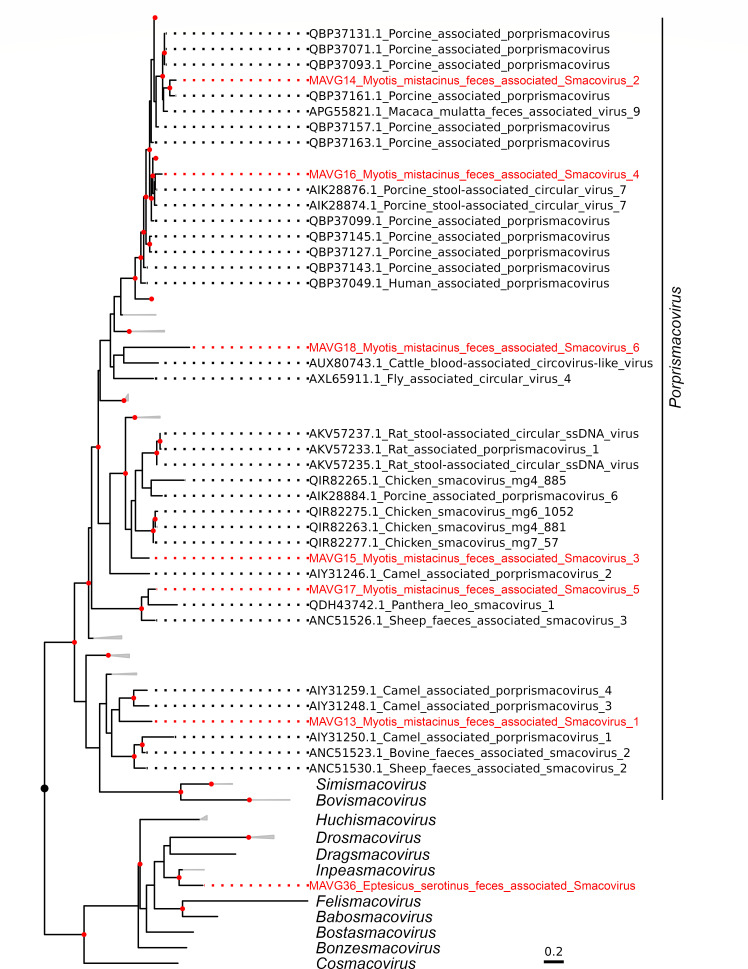
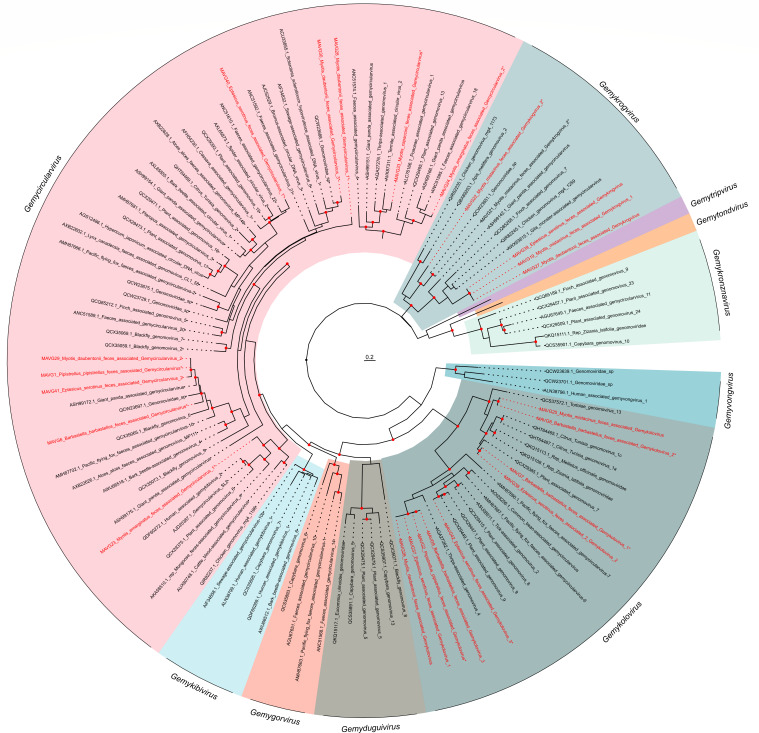
Bats are natural hosts of multiple viruses, many of which have clear zoonotic potential. The search for emerging viruses has been aided by the implementation of metagenomic tools, which have also enabled the detection of unprecedented viral diversity. Currently, this search is mainly focused on RNA viruses, which are largely over-represented in databases. To compensate for this research bias, we analyzed fecal samples from 189 Spanish bats belonging to 22 different species using viral metagenomics. This allowed us to identify 52 complete or near-complete viral genomes belonging to the families Adenoviridae, Circoviridae, Genomoviridae, Papillomaviridae, Parvoviridae, Polyomaviridae and Smacoviridae. Of these, 30 could constitute new species, doubling the number of viruses currently described in Europe. These findings open the door to a more thorough analysis of bat DNA viruses and their zoonotic potential.
Importance: Metagenomics has become a fundamental tool to characterize the global virosphere, allowing us not only to understand the existing viral diversity and its ecological implications but also to identify new and emerging viruses. RNA viruses have a higher zoonotic potential, but this risk is also present for some DNA virus families. In our study, we analyzed the DNA fraction of fecal samples from 22 Spanish bat species, identifying 52 complete or near-complete genomes of different viral families with zoonotic potential. This doubles the number of genomes currently described in Europe. Metagenomic data often produce partial genomes that can be difficult to analyze. Our work, however, has characterized a large number of complete genomes, thus facilitating their taxonomic classification and enabling different analyses to be carried out to evaluate their zoonotic potential. For example, recombination studies are relevant since this phenomenon could play a major role in cross-species transmission.
Keywords: DNA viruses; bat viruses; metagenomics; viral emergence; viromics; zoonotic viruses.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Complete Genomes of DNA Viruses in Fecal Samples from Small Terrestrial Mammals in Spain.Viruses. 2024 Dec 5;16(12):1885. doi: 10.3390/v16121885. Viruses. 2024. PMID: 39772193 Free PMC article.
-
Genetic diversity and cross-species transmissibility of bat-associated picornaviruses from Spain.Virol J. 2024 Aug 22;21(1):193. doi: 10.1186/s12985-024-02456-1. Virol J. 2024. PMID: 39175061 Free PMC article.
-
Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses.J Virol. 2010 Jul;84(14):6955-65. doi: 10.1128/JVI.00501-10. Epub 2010 May 12. J Virol. 2010. PMID: 20463061 Free PMC article.
-
Bats as Viral Reservoirs.Annu Rev Virol. 2016 Sep 29;3(1):77-99. doi: 10.1146/annurev-virology-110615-042203. Epub 2016 Aug 22. Annu Rev Virol. 2016. PMID: 27578437 Review.
-
The Virome of Bats Inhabiting Brazilian Biomes: Knowledge Gaps and Biases towards Zoonotic Viruses.Microbiol Spectr. 2023 Feb 14;11(1):e0407722. doi: 10.1128/spectrum.04077-22. Epub 2023 Jan 10. Microbiol Spectr. 2023. PMID: 36625641 Free PMC article. Review.
Cited by
-
Phylogenetic evidence supporting the nonenveloped nature of hepadnavirus ancestors.Proc Natl Acad Sci U S A. 2024 Nov 5;121(45):e2415631121. doi: 10.1073/pnas.2415631121. Epub 2024 Oct 29. Proc Natl Acad Sci U S A. 2024. PMID: 39471221 Free PMC article.
-
Identification and characterization of novel bat coronaviruses in Spain.PLoS Pathog. 2025 Aug 4;21(8):e1013371. doi: 10.1371/journal.ppat.1013371. eCollection 2025 Aug. PLoS Pathog. 2025. PMID: 40758759 Free PMC article.
-
Anogenital papillomatosis associated with a novel papillomavirus in a grey-headed flying fox.J Vet Diagn Invest. 2025 May 24:10406387251341935. doi: 10.1177/10406387251341935. Online ahead of print. J Vet Diagn Invest. 2025. PMID: 40411201 Free PMC article.
-
Complete Genomes of DNA Viruses in Fecal Samples from Small Terrestrial Mammals in Spain.Viruses. 2024 Dec 5;16(12):1885. doi: 10.3390/v16121885. Viruses. 2024. PMID: 39772193 Free PMC article.
-
Characterization of Three Novel Papillomavirus Genomes in Vampire Bats (Desmodus rotundus).Animals (Basel). 2024 Dec 14;14(24):3604. doi: 10.3390/ani14243604. Animals (Basel). 2024. PMID: 39765508 Free PMC article.
References
-
- Bolatti EM, Zorec TM, Montani ME, Hošnjak L, Chouhy D, Viarengo G, Casal PE, Barquez RM, Poljak M, Giri AA. 2020. A preliminary study of the virome of the South American free-tailed bats (Tadarida brasiliensis) and identification of two novel mammalian viruses. Viruses 12:422. doi: 10.3390/v12040422 - DOI - PMC - PubMed
-
- Van Brussel K, Mahar JE, Ortiz-Baez AS, Carrai M, Spielman D, Boardman WSJ, Baker ML, Beatty JA, Geoghegan JL, Barrs VR, Holmes EC. 2022. Faecal virome of the Australian grey-headed flying fox from urban/suburban environments contains novel coronaviruses, retroviruses and sapoviruses. Virology 576:42–51. doi: 10.1016/j.virol.2022.09.002 - DOI - PubMed
-
- Wu Z, Yang L, Ren X, He G, Zhang J, Yang J, Qian Z, Dong J, Sun L, Zhu Y, Du J, Yang F, Zhang S, Jin Q. 2016. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J 10:609–620. doi: 10.1038/ismej.2015.138 - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
