

GAPS: a geometric attention-based network for peptide binding site identification by the transfer learning approach
- PMID: 38990514
- PMCID: PMC11238429
- DOI: 10.1093/bib/bbae297
GAPS: a geometric attention-based network for peptide binding site identification by the transfer learning approach
Abstract
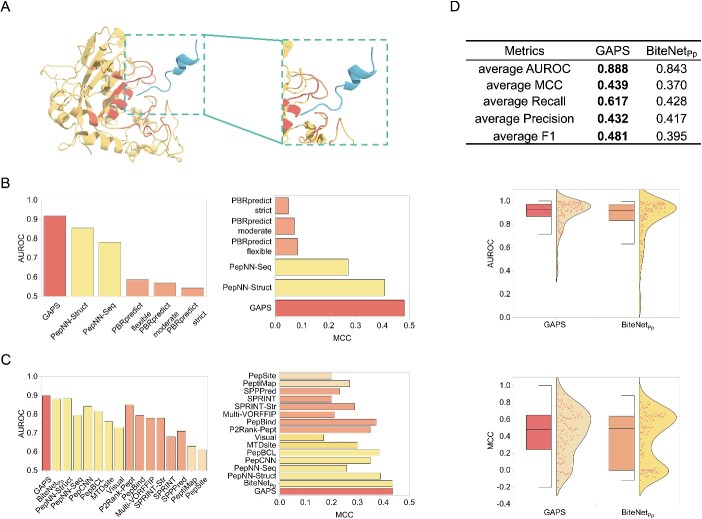
Protein-peptide interactions (PPepIs) are vital to understanding cellular functions, which can facilitate the design of novel drugs. As an essential component in forming a PPepI, protein-peptide binding sites are the basis for understanding the mechanisms involved in PPepIs. Therefore, accurately identifying protein-peptide binding sites becomes a critical task. The traditional experimental methods for researching these binding sites are labor-intensive and time-consuming, and some computational tools have been invented to supplement it. However, these computational tools have limitations in generality or accuracy due to the need for ligand information, complex feature construction, or their reliance on modeling based on amino acid residues. To deal with the drawbacks of these computational algorithms, we describe a geometric attention-based network for peptide binding site identification (GAPS) in this work. The proposed model utilizes geometric feature engineering to construct atom representations and incorporates multiple attention mechanisms to update relevant biological features. In addition, the transfer learning strategy is implemented for leveraging the protein-protein binding sites information to enhance the protein-peptide binding sites recognition capability, taking into account the common structure and biological bias between proteins and peptides. Consequently, GAPS demonstrates the state-of-the-art performance and excellent robustness in this task. Moreover, our model exhibits exceptional performance across several extended experiments including predicting the apo protein-peptide, protein-cyclic peptide and the AlphaFold-predicted protein-peptide binding sites. These results confirm that the GAPS model is a powerful, versatile, stable method suitable for diverse binding site predictions.
Keywords: attention mechanism; binding sites; geometric deep learning; transfer learning.
© The Author(s) 2024. Published by Oxford University Press.
Figures





Similar articles
-
DP-site: A dual deep learning-based method for protein-peptide interaction site prediction.Methods. 2024 Sep;229:17-29. doi: 10.1016/j.ymeth.2024.06.001. Epub 2024 Jun 12. Methods. 2024. PMID: 38871095
-
Turbocharging protein binding site prediction with geometric attention, inter-resolution transfer learning, and homology-based augmentation.BMC Bioinformatics. 2024 Sep 20;25(1):306. doi: 10.1186/s12859-024-05923-2. BMC Bioinformatics. 2024. PMID: 39304807 Free PMC article.
-
A deep-learning framework for multi-level peptide-protein interaction prediction.Nat Commun. 2021 Sep 15;12(1):5465. doi: 10.1038/s41467-021-25772-4. Nat Commun. 2021. PMID: 34526500 Free PMC article.
-
A comprehensive survey on protein-ligand binding site prediction.Curr Opin Struct Biol. 2024 Jun;86:102793. doi: 10.1016/j.sbi.2024.102793. Epub 2024 Mar 5. Curr Opin Struct Biol. 2024. PMID: 38447285 Review.
-
Research progress on prediction of RNA-protein binding sites in the past five years.Anal Biochem. 2024 Aug;691:115535. doi: 10.1016/j.ab.2024.115535. Epub 2024 Apr 20. Anal Biochem. 2024. PMID: 38643894 Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
