

De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome
- PMID: 38991538
- PMCID: PMC11338827
- DOI: 10.1038/s41586-024-07773-7
De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome
Abstract
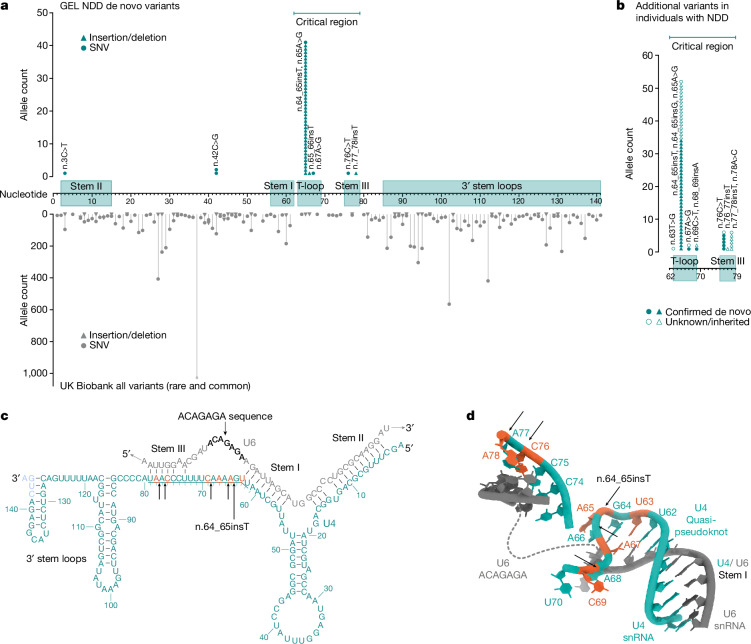
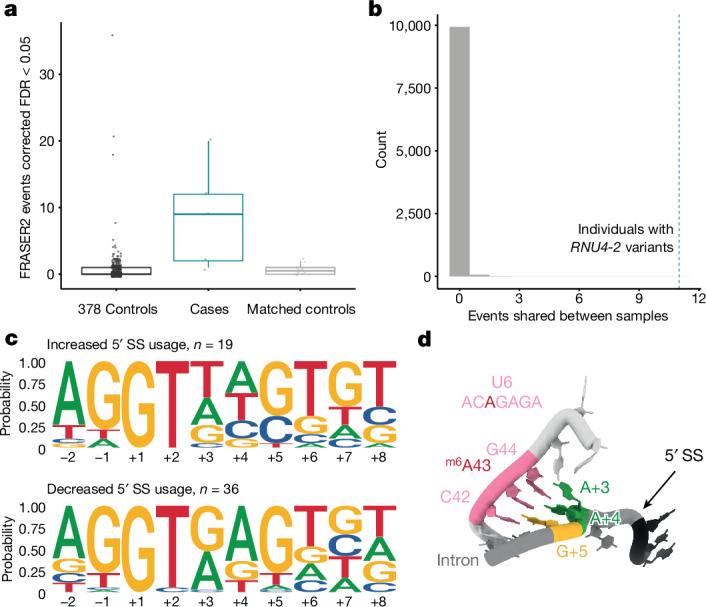
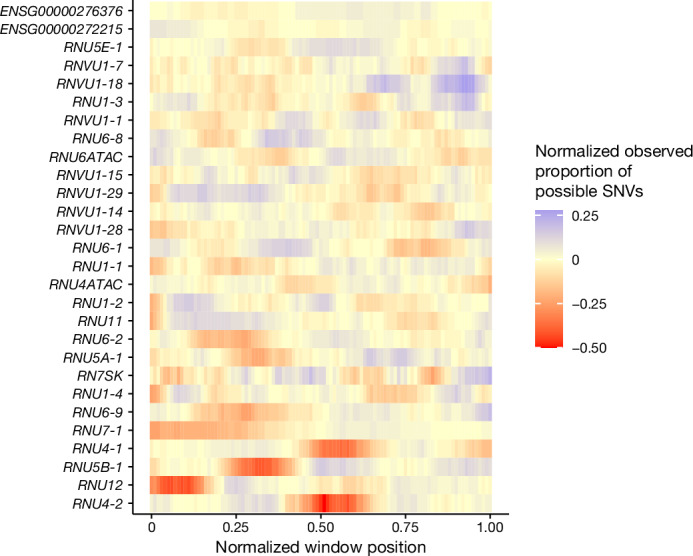
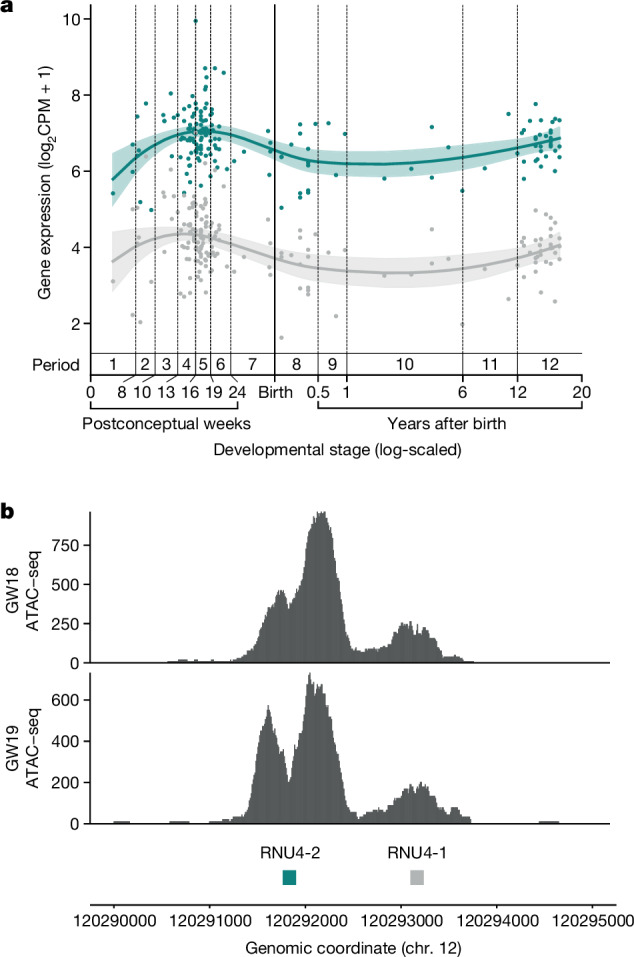
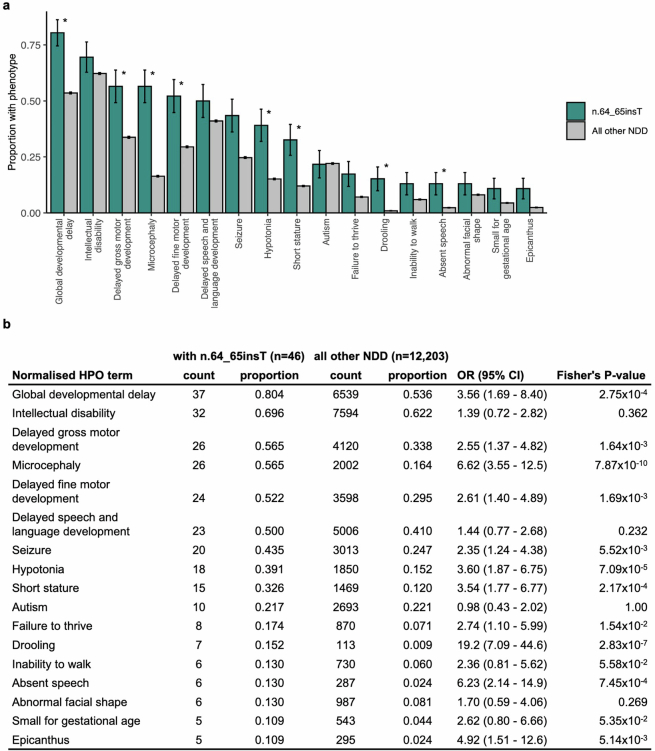
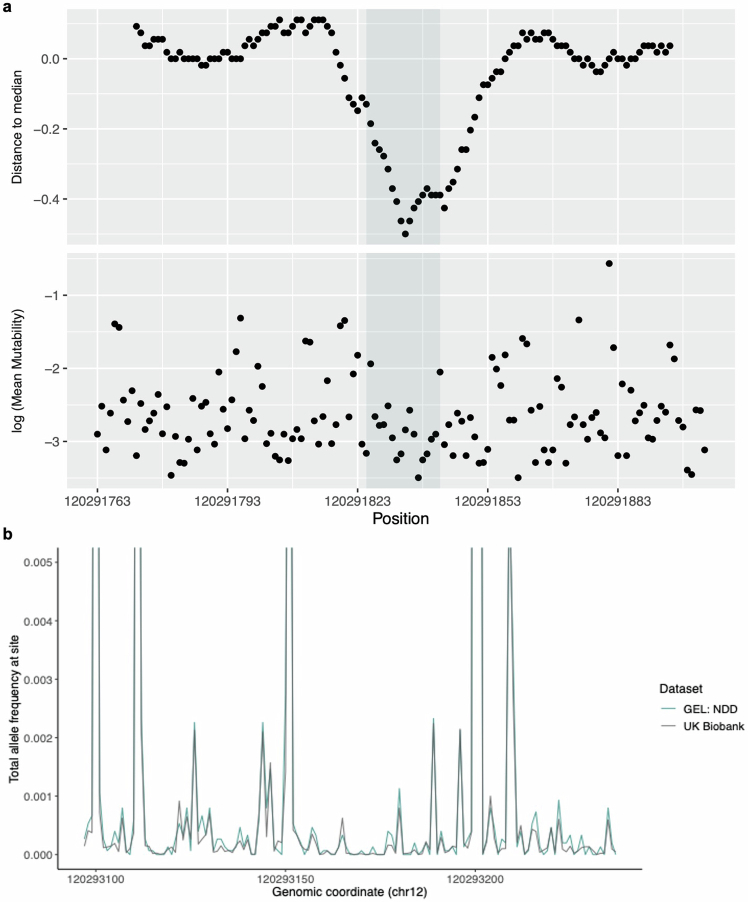
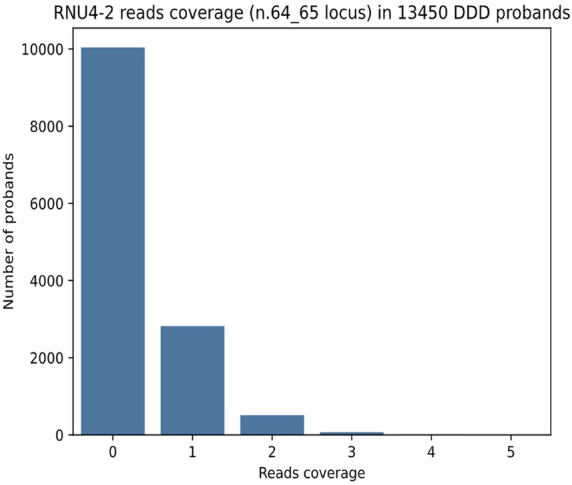
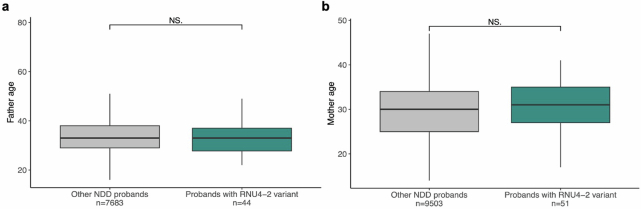
Around 60% of individuals with neurodevelopmental disorders (NDD) remain undiagnosed after comprehensive genetic testing, primarily of protein-coding genes1. Large genome-sequenced cohorts are improving our ability to discover new diagnoses in the non-coding genome. Here we identify the non-coding RNA RNU4-2 as a syndromic NDD gene. RNU4-2 encodes the U4 small nuclear RNA (snRNA), which is a critical component of the U4/U6.U5 tri-snRNP complex of the major spliceosome2. We identify an 18 base pair region of RNU4-2 mapping to two structural elements in the U4/U6 snRNA duplex (the T-loop and stem III) that is severely depleted of variation in the general population, but in which we identify heterozygous variants in 115 individuals with NDD. Most individuals (77.4%) have the same highly recurrent single base insertion (n.64_65insT). In 54 individuals in whom it could be determined, the de novo variants were all on the maternal allele. We demonstrate that RNU4-2 is highly expressed in the developing human brain, in contrast to RNU4-1 and other U4 homologues. Using RNA sequencing, we show how 5' splice-site use is systematically disrupted in individuals with RNU4-2 variants, consistent with the known role of this region during spliceosome activation. Finally, we estimate that variants in this 18 base pair region explain 0.4% of individuals with NDD. This work underscores the importance of non-coding genes in rare disorders and will provide a diagnosis to thousands of individuals with NDD worldwide.
© 2024. The Author(s).
Conflict of interest statement
N.W. receives research funding from Novo Nordisk and has consulted for ArgoBio studio. S.J.S. receives research funding from BioMarin Pharmaceutical. A.O.’D.-L. is on the scientific advisory board for Congenica, was a paid consultant for Tome Biosciences, Ono Pharma USA Inc. and at present for Addition Therapeutics, and received reagents from PacBio to support rare disease research. H.L.R. has received support from Illumina and Microsoft to support rare disease gene discovery and diagnosis. M.W. has consulted for Illumina and Sanofi and received speaking honoraria from Illumina and GeneDx. S.B.M. is an advisor for BioMarin, Myome and Tenaya Therapeutics. S.M.S. has received honoraria for educational events or advisory boards from Angelini Pharma, Biocodex, Eisai, Zogenix/UCB and institutional contributions for advisory boards, educational events or consultancy work from Eisai, Jazz/GW Pharma, Stoke Therapeutics, Takeda, UCB and Zogenix. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics Laboratories. J.M.M.H. is a full-time employee of Novo Nordisk and holds shares in Novo Nordisk A/S. D.G.M. is a paid consultant for GlaxoSmithKline, Insitro and Overtone Therapeutics and receives research support from Microsoft. All other authors declare no competing interests.
Figures

Update of
-
De novo variants in the non-coding spliceosomal snRNA gene RNU4-2 are a frequent cause of syndromic neurodevelopmental disorders.medRxiv [Preprint]. 2024 Apr 9:2024.04.07.24305438. doi: 10.1101/2024.04.07.24305438. medRxiv. 2024. Update in: Nature. 2024 Aug;632(8026):832-840. doi: 10.1038/s41586-024-07773-7. PMID: 38645094 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- UM1 TR004409/TR/NCATS NIH HHS/United States
- 220134/WT_/Wellcome Trust/United Kingdom
- U01 HG007709/HG/NHGRI NIH HHS/United States
- U01 HG011762/HG/NHGRI NIH HHS/United States
- U01 HG011755/HG/NHGRI NIH HHS/United States
- U01 HG010217/HG/NHGRI NIH HHS/United States
- R21 HG012397/HG/NHGRI NIH HHS/United States
- U01 HG007942/HG/NHGRI NIH HHS/United States
- U01 NS134358/NS/NINDS NIH HHS/United States
- U24 HG011746/HG/NHGRI NIH HHS/United States
- R01 MH129751/MH/NIMH NIH HHS/United States
- U01 NS134348/NS/NINDS NIH HHS/United States
- R01 MH122681/MH/NIMH NIH HHS/United States
- R01 HG009141/HG/NHGRI NIH HHS/United States
- K23 AR083505/AR/NIAMS NIH HHS/United States
- U54 NS115052/NS/NINDS NIH HHS/United States
- U24 NS131172/NS/NINDS NIH HHS/United States
- U01 HG011745/HG/NHGRI NIH HHS/United States
- U01 NS106845/NS/NINDS NIH HHS/United States
- P50 HD103555/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
