

Chasing Sequencing Perfection: Marching Toward Higher Accuracy and Lower Costs
- PMID: 38991976
- PMCID: PMC11423848
- DOI: 10.1093/gpbjnl/qzae024
Chasing Sequencing Perfection: Marching Toward Higher Accuracy and Lower Costs
Abstract
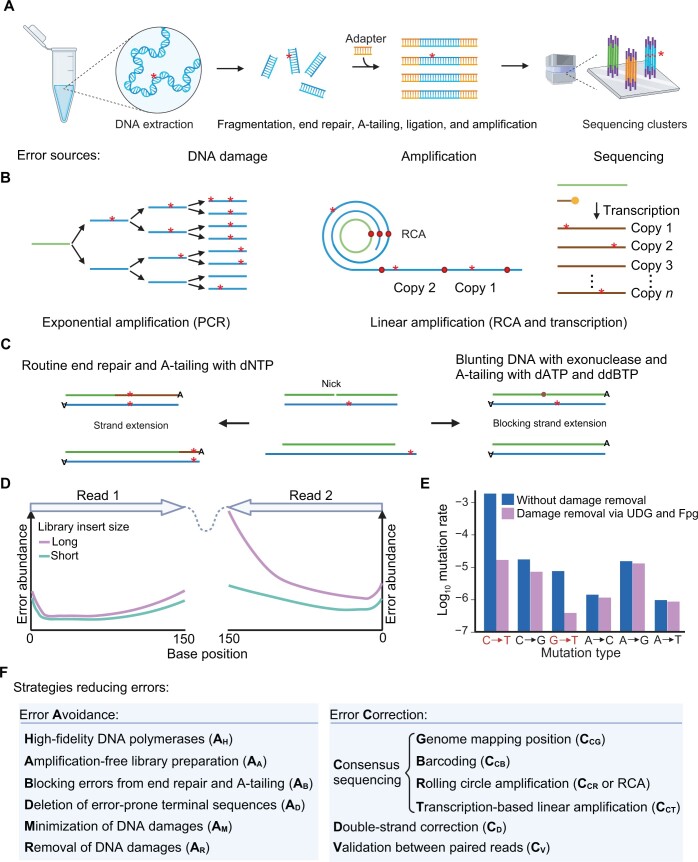
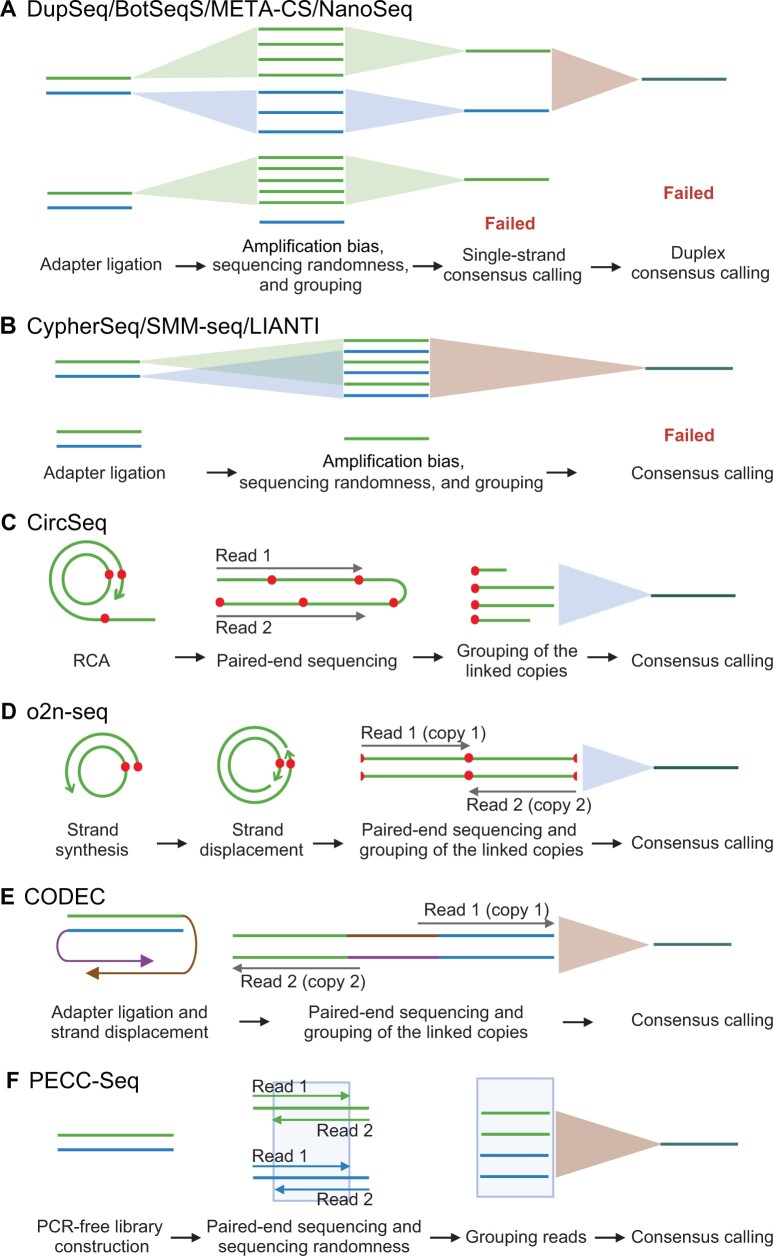
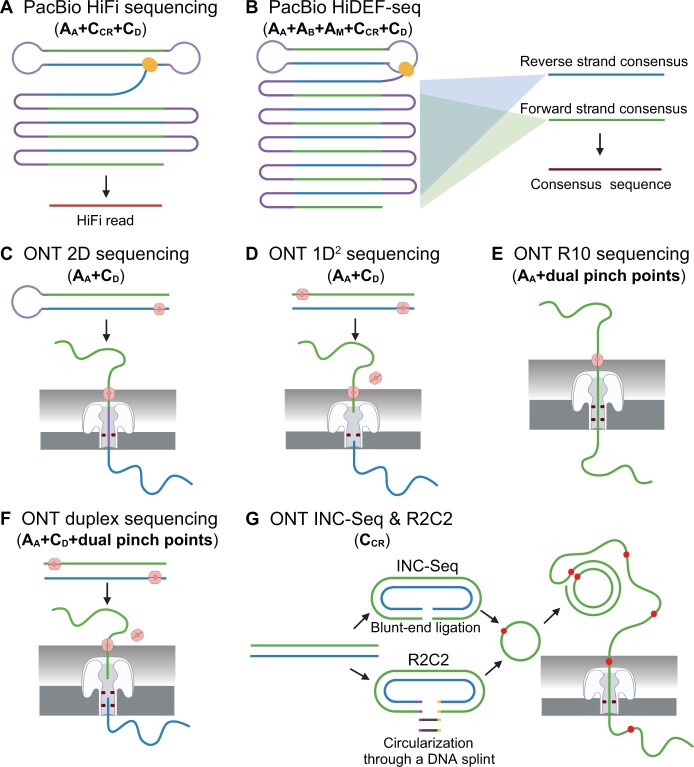
Next-generation sequencing (NGS), represented by Illumina platforms, has been an essential cornerstone of basic and applied research. However, the sequencing error rate of 1 per 1000 bp (10-3) represents a serious hurdle for research areas focusing on rare mutations, such as somatic mosaicism or microbe heterogeneity. By examining the high-fidelity sequencing methods developed in the past decade, we summarized three major factors underlying errors and the corresponding 12 strategies mitigating these errors. We then proposed a novel framework to classify 11 preexisting representative methods according to the corresponding combinatory strategies and identified three trends that emerged during methodological developments. We further extended this analysis to eight long-read sequencing methods, emphasizing error reduction strategies. Finally, we suggest two promising future directions that could achieve comparable or even higher accuracy with lower costs in both NGS and long-read sequencing.
Keywords: Consensus sequencing; High-fidelity sequencing; Rare mutation; Sequencing error; Single-molecule sequencing.
© The Author(s) 2024. Published by Oxford University Press and Science Press on behalf of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China.
Conflict of interest statement
The authors have declared no competing interests.
Figures
Similar articles
-
Whole-genome and targeted sequencing of drug-resistant Mycobacterium tuberculosis on the iSeq100 and MiSeq: A performance, ease-of-use, and cost evaluation.PLoS Med. 2019 Apr 30;16(4):e1002794. doi: 10.1371/journal.pmed.1002794. eCollection 2019 Apr. PLoS Med. 2019. PMID: 31039166 Free PMC article.
-
Is the $1000 Genome as Near as We Think? A Cost Analysis of Next-Generation Sequencing.Clin Chem. 2016 Nov;62(11):1458-1464. doi: 10.1373/clinchem.2016.258632. Epub 2016 Sep 14. Clin Chem. 2016. PMID: 27630156
-
Pseudo-Sanger sequencing: massively parallel production of long and near error-free reads using NGS technology.BMC Genomics. 2013 Oct 17;14(1):711. doi: 10.1186/1471-2164-14-711. BMC Genomics. 2013. PMID: 24134808 Free PMC article.
-
Next-generation sequencing technologies: An overview.Hum Immunol. 2021 Nov;82(11):801-811. doi: 10.1016/j.humimm.2021.02.012. Epub 2021 Mar 19. Hum Immunol. 2021. PMID: 33745759 Review.
-
Ten years of next-generation sequencing technology.Trends Genet. 2014 Sep;30(9):418-26. doi: 10.1016/j.tig.2014.07.001. Epub 2014 Aug 6. Trends Genet. 2014. PMID: 25108476 Review.
Cited by
-
5-Hydroxymethylcytosine modifications in circulating cell-free DNA: frontiers of cancer detection, monitoring, and prognostic evaluation.Biomark Res. 2025 Mar 7;13(1):39. doi: 10.1186/s40364-025-00751-9. Biomark Res. 2025. PMID: 40055844 Free PMC article. Review.
-
Unlocking the Potential of Metagenomics with the PacBio High-Fidelity Sequencing Technology.Microorganisms. 2024 Dec 2;12(12):2482. doi: 10.3390/microorganisms12122482. Microorganisms. 2024. PMID: 39770685 Free PMC article. Review.
-
Advancing genome-based precision medicine: a review on machine learning applications for rare genetic disorders.Brief Bioinform. 2025 Jul 2;26(4):bbaf329. doi: 10.1093/bib/bbaf329. Brief Bioinform. 2025. PMID: 40668553 Free PMC article. Review.
References
-
- Shendure J, Ji H.. Next-generation DNA sequencing. Nat Biotechnol 2008;26:1135–45. - PubMed
-
- Ewing B, Green P.. Base-calling of automated sequencer traces using phred. II. error probabilities. Genome Res 1998;8:186–94. - PubMed
-
- Ewing B, Hillier L, Wendl MC, Green P.. Base-calling of automated sequencer traces using phred. I. accuracy assessment. Genome Res 1998;8:175–85. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
