

Biliary atresia
- PMID: 38992031
- PMCID: PMC11956545
- DOI: 10.1038/s41572-024-00533-x
Biliary atresia
Abstract
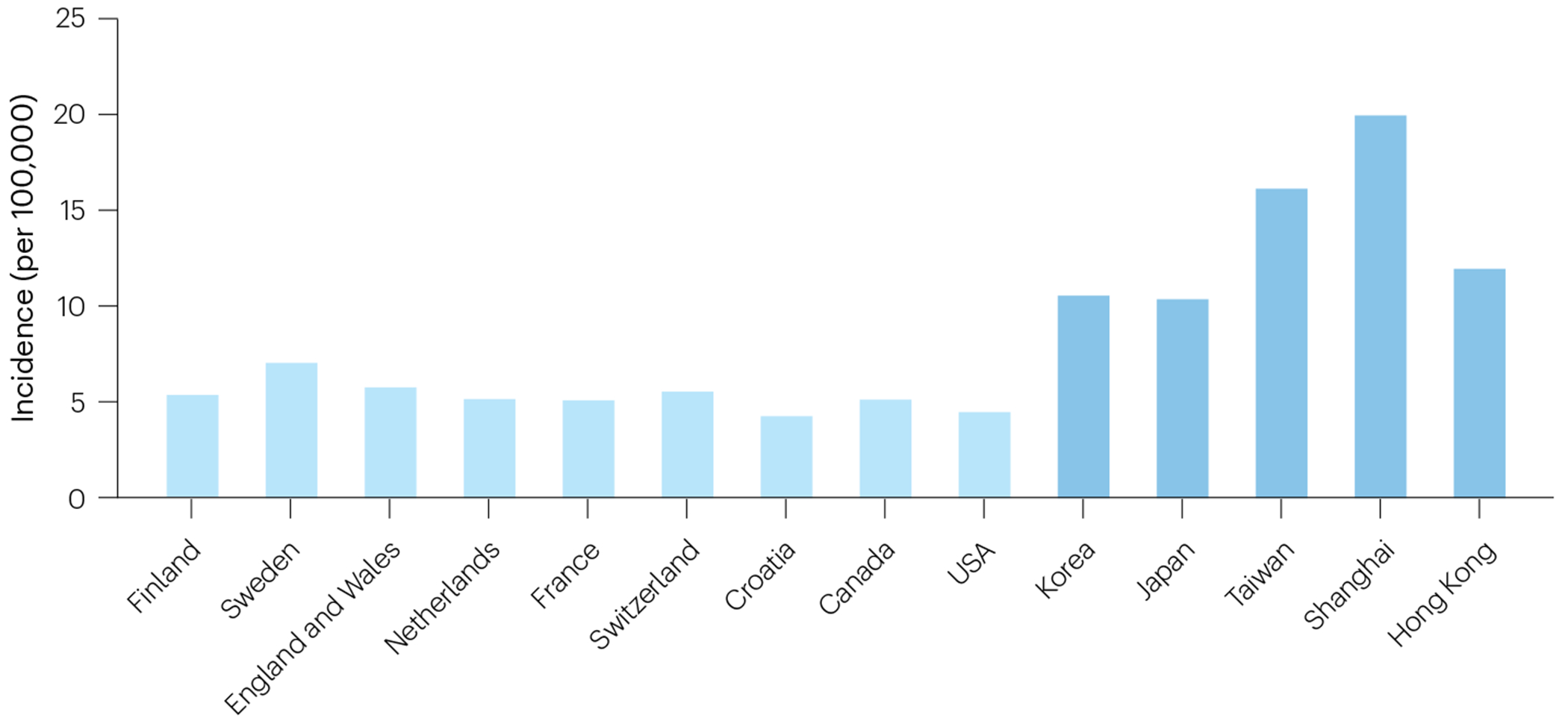
Biliary atresia (BA) is a progressive inflammatory fibrosclerosing disease of the biliary system and a major cause of neonatal cholestasis. It affects 1:5,000-20,000 live births, with the highest incidence in Asia. The pathogenesis is still unknown, but emerging research suggests a role for ciliary dysfunction, redox stress and hypoxia. The study of the underlying mechanisms can be conceptualized along the likely prenatal timing of an initial insult and the distinction between the injury and prenatal and postnatal responses to injury. Although still speculative, these emerging concepts, new diagnostic tools and early diagnosis might enable neoadjuvant therapy (possibly aimed at oxidative stress) before a Kasai portoenterostomy (KPE). This is particularly important, as timely KPE restores bile flow in only 50-75% of patients of whom many subsequently develop cholangitis, portal hypertension and progressive fibrosis; 60-75% of patients require liver transplantation by the age of 18 years. Early diagnosis, multidisciplinary management, centralization of surgery and optimized interventions for complications after KPE lead to better survival. Postoperative corticosteroid use has shown benefits, whereas the role of other adjuvant therapies remains to be evaluated. Continued research to better understand disease mechanisms is necessary to develop innovative treatments, including adjuvant therapies targeting the immune response, regenerative medicine approaches and new clinical tests to improve patient outcomes.
© 2024. Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures







References
-
- Hartley JL, Davenport M & Kelly DA Biliary atresia. Lancet 374, 1704–1713 (2009). - PubMed
-
-
Chung PHY, Zheng S & Tam PKH Biliary atresia: East versus West. Semin. Pediatr. Surg 29, 150950 (2020).
A review summarizing the geographical differences in BA aetiology and treatment.
-
-
-
Davenport M, Muntean A & Hadzic N Biliary atresia: clinical phenotypes and aetiological heterogeneity. J. Clin. Med 10, 5675 (2021).
A description of different BA phenotypes and associated malformations.
-
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
