

INDELpred: Improving the prediction and interpretation of indel pathogenicity within the clinical genome
- PMID: 38993112
- PMCID: PMC11321314
- DOI: 10.1016/j.xhgg.2024.100325
INDELpred: Improving the prediction and interpretation of indel pathogenicity within the clinical genome
Abstract
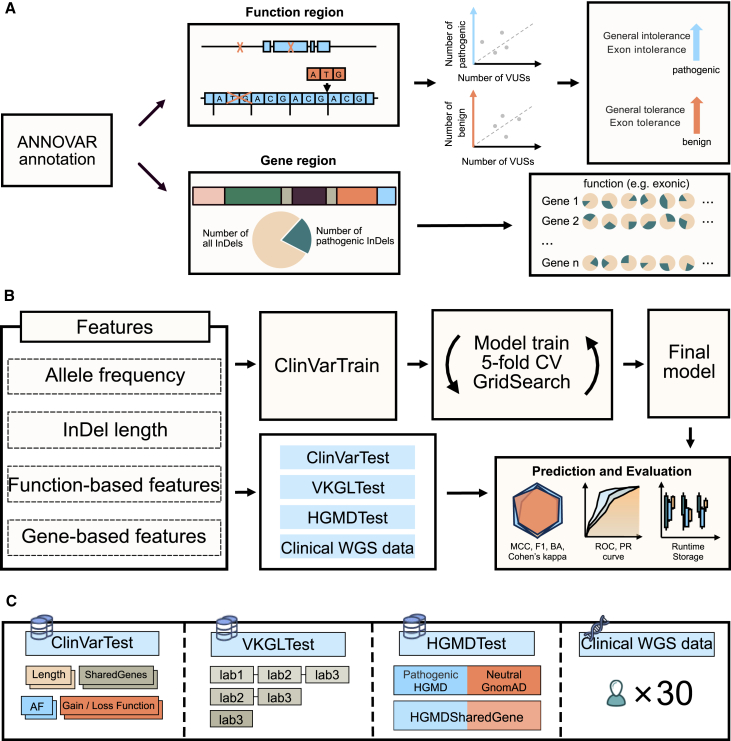
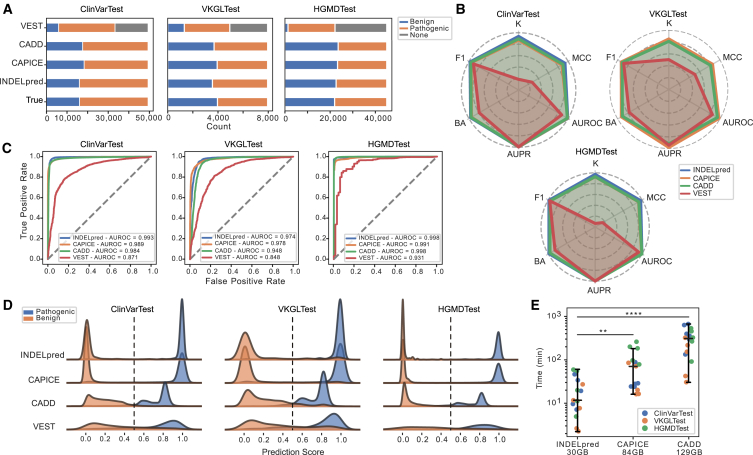
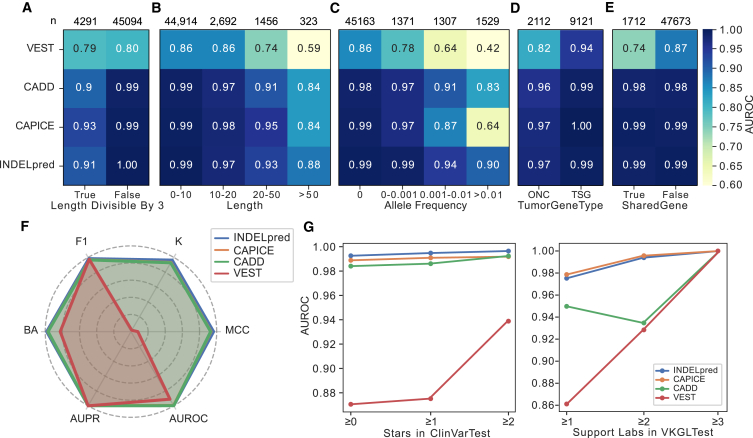
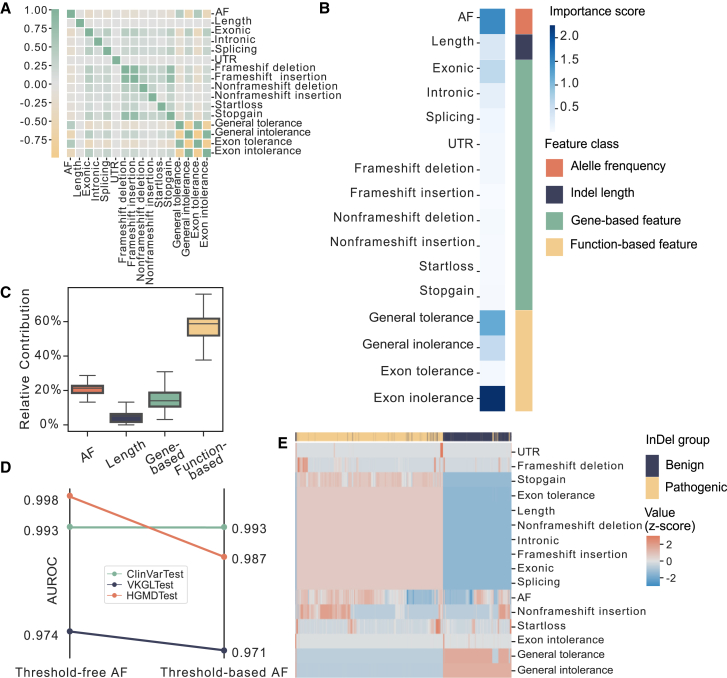
Small insertions and deletions (indels) are critical yet challenging genetic variations with significant clinical implications. However, the identification of pathogenic indels from neutral variants in clinical contexts remains an understudied problem. Here, we developed INDELpred, a machine-learning-based predictive model for discerning pathogenic from benign indels. INDELpred was established based on key features, including allele frequency, indel length, function-based features, and gene-based features. A set of comprehensive evaluation analyses demonstrated that INDELpred exhibited superior performance over competing methods in terms of computational efficiency and prediction accuracy. Importantly, INDELpred highlighted the crucial role of function-based features in identifying pathogenic indels, with a clear interpretability of the features in understanding the disease-causing variants. We envisage INDELpred as a desirable tool for the detection of pathogenic indels within large-scale genomic datasets, thereby enhancing the precision of genetic diagnoses in clinical settings.
Keywords: InDel; clinical genomics; machine learning; pathogenicity prediction; whole genome sequencing.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

Similar articles
-
An integrative approach to predicting the functional effects of small indels in non-coding regions of the human genome.BMC Bioinformatics. 2017 Oct 6;18(1):442. doi: 10.1186/s12859-017-1862-y. BMC Bioinformatics. 2017. PMID: 28985712 Free PMC article.
-
Detecting Somatic Insertions/Deletions (Indels) Using Tumor RNA-Seq Data.Methods Mol Biol. 2024;2812:235-242. doi: 10.1007/978-1-0716-3886-6_13. Methods Mol Biol. 2024. PMID: 39068366
-
Tool evaluation for the detection of variably sized indels from next generation whole genome and targeted sequencing data.PLoS Comput Biol. 2022 Feb 17;18(2):e1009269. doi: 10.1371/journal.pcbi.1009269. eCollection 2022 Feb. PLoS Comput Biol. 2022. PMID: 35176018 Free PMC article.
-
Small Insertions and Deletions Drive Genomic Plasticity during Adaptive Evolution of Yersinia pestis.Microbiol Spectr. 2022 Jun 29;10(3):e0224221. doi: 10.1128/spectrum.02242-21. Epub 2022 Apr 19. Microbiol Spectr. 2022. PMID: 35438532 Free PMC article. Review.
-
Biomedical informatics and machine learning for clinical genomics.Hum Mol Genet. 2018 May 1;27(R1):R29-R34. doi: 10.1093/hmg/ddy088. Hum Mol Genet. 2018. PMID: 29566172 Free PMC article. Review.
References
-
- The 100,000 Genomes Project Pilot Investigators. Smedley D., Smith K.R., Martin A., Thomas E.A., McDonagh E.M., Cipriani V., Ellingford J.M., Arno G., Tucci A., et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care — Preliminary Report. N. Engl. J. Med. 2021;385:1868–1880. doi: 10.1056/NEJMoa2035790. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
