

Characterization and genomic analysis of a lytic Stenotrophomonas maltophilia short-tailed phage A1432 revealed a new genus of the family Mesyanzhinovviridae
- PMID: 38993489
- PMCID: PMC11236537
- DOI: 10.3389/fmicb.2024.1400700
Characterization and genomic analysis of a lytic Stenotrophomonas maltophilia short-tailed phage A1432 revealed a new genus of the family Mesyanzhinovviridae
Abstract
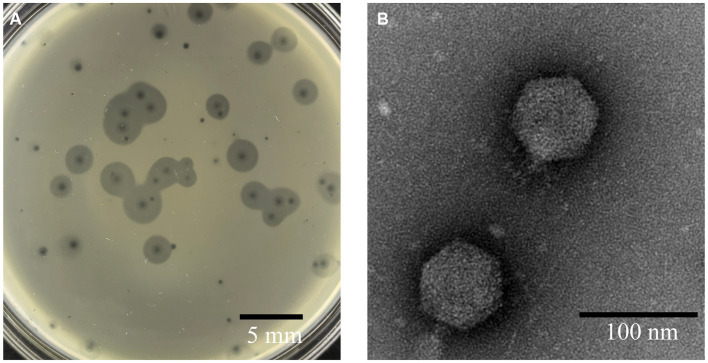
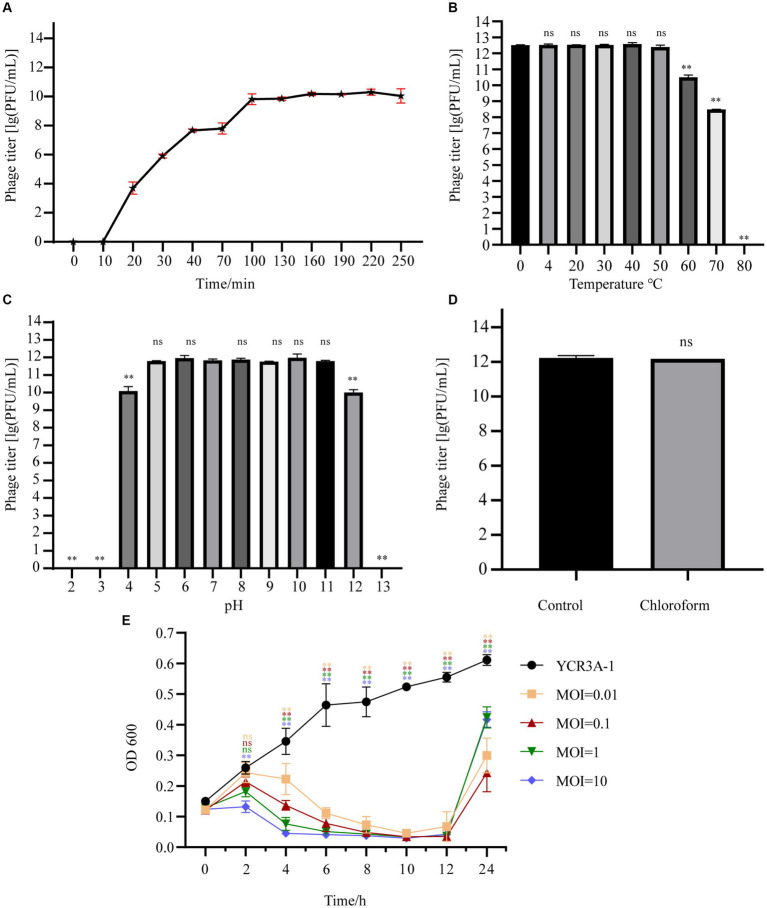
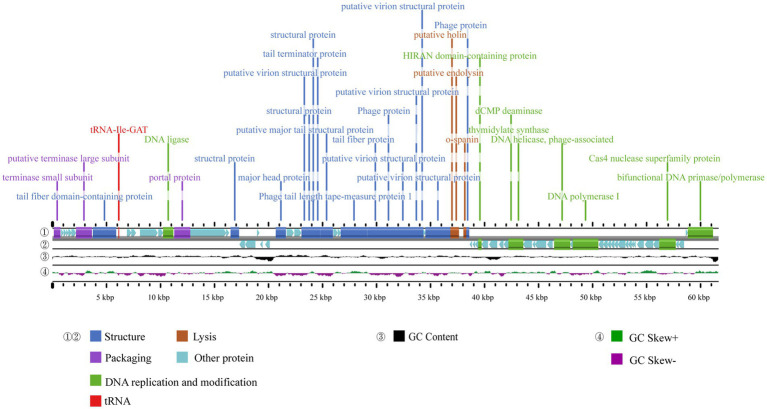
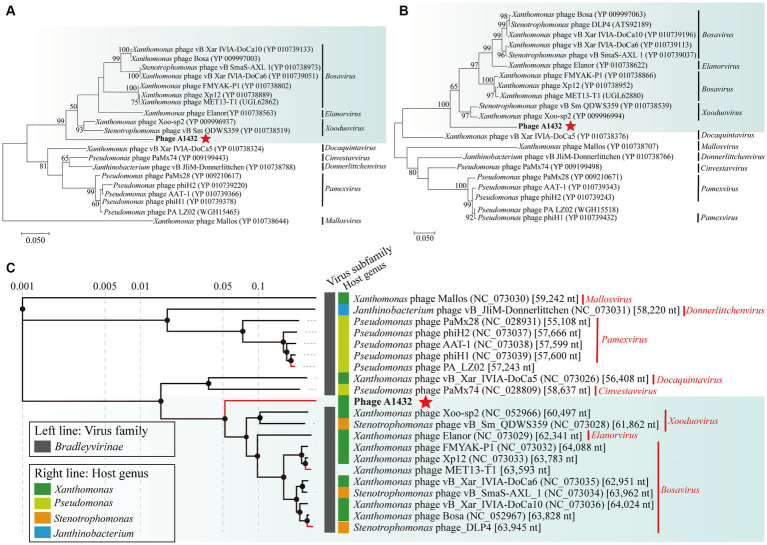
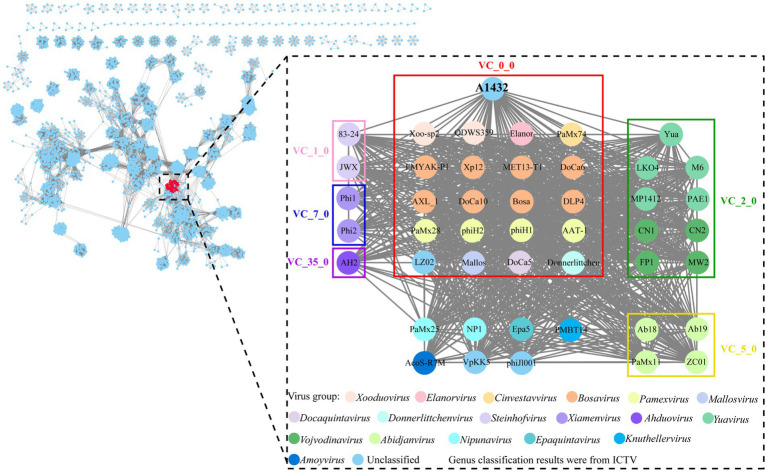
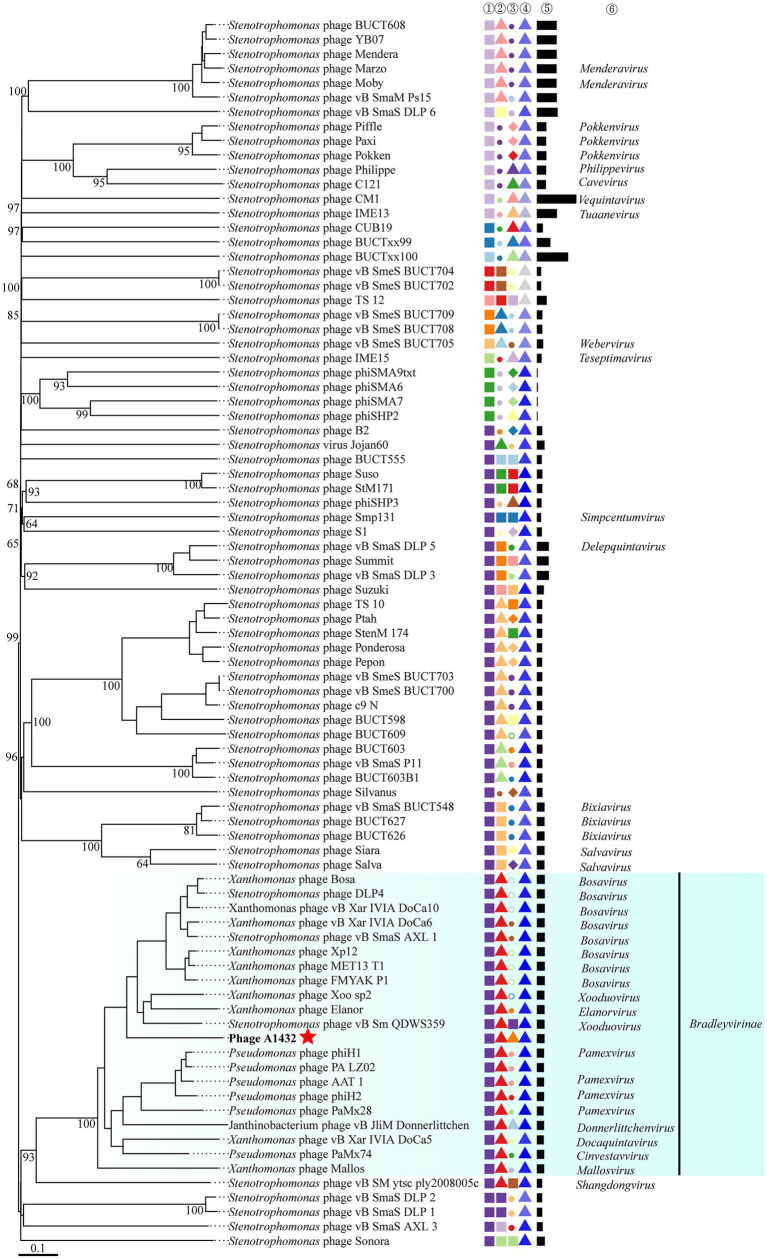
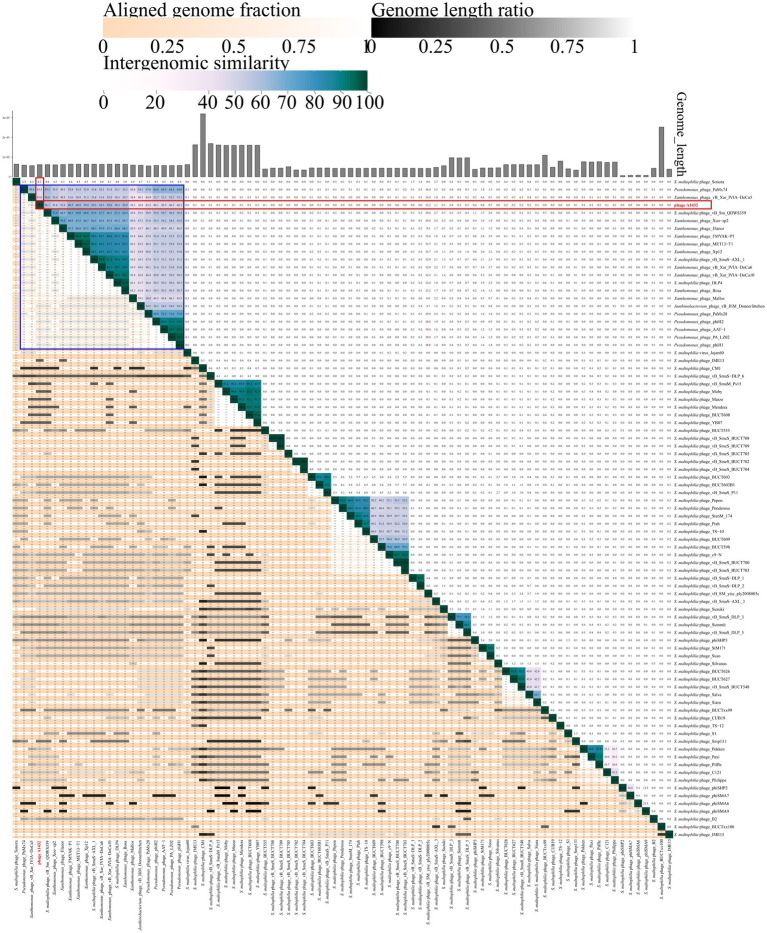
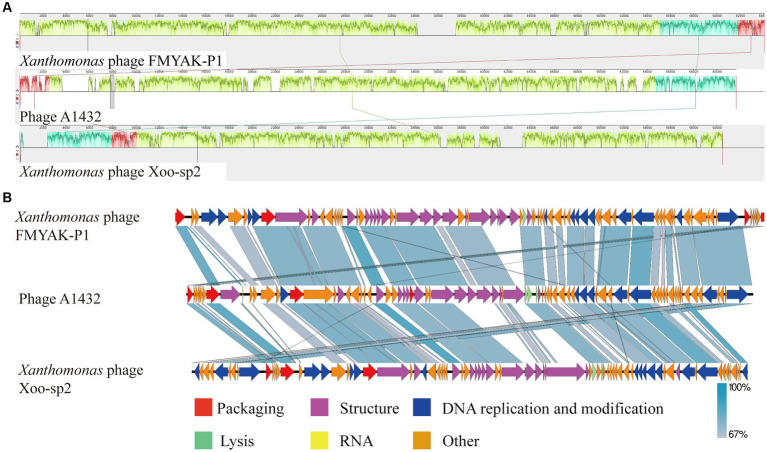
Stenotrophomonas maltophilia (S. maltophilia) is an emerging opportunistic pathogen that exhibits resistant to a majority of commonly used antibiotics. Phages have the potential to serve as an alternative treatment for S. maltophilia infections. In this study, a lytic phage, A1432, infecting S. maltophilia YCR3A-1, was isolated and characterized from a karst cave. Transmission electron microscopy revealed that phage A1432 possesses an icosahedral head and a shorter tail. Phage A1432 demonstrated a narrow host range, with an optimal multiplicity of infection of 0.1. The one-step growth curve indicated a latent time of 10 min, a lysis period of 90 min, a burst size of 43.2 plaque-forming units per cell. In vitro bacteriolytic activity test showed that phage A1432 was capable to inhibit the growth of S. maltophilia YCR3A-1 in an MOI-dependent manner after 2 h of co-culture. BLASTn analysis showed that phage A1432 genome shares the highest similarity (81.46%) with Xanthomonas phage Xoo-sp2 in the NCBI database, while the query coverage was only 37%. The phage contains double-stranded DNA with a genome length of 61,660 bp and a GC content of 61.92%. It is predicted to have 79 open reading frames and one tRNA, with no virulence or antibiotic resistance genes. Phylogenetic analysis using terminase large subunit and DNA polymerase indicated that phage A1432 clustered with members of the Bradleyvirinae subfamily but diverged into a distinct branch. Further phylogenetic comparison analysis using Average Nucleotide Identity, proteomic phylogenetic analysis, genomic network analysis confirmed that phage A1432 belongs to a novel genus within the Bradleyvirinae subfamily, Mesyanzhinovviridae family. Additionally, phylogenetic analysis of the so far isolated S. maltophilia phages revealed significant genetic diversity among these phages. The results of this research will contribute valuable information for further studies on their morphological and genetic diversity, will aid in elucidating the evolutionary mechanisms that give rise to them.
Keywords: Bradleyvirinae; Mesyanzhinovviridae; Stenotrophomonas maltophilia; karst cave; lytic phage; new genus; phage.
Copyright © 2024 Li, Xu, Yang, Yang, Wu, Li, Yang, Fang, Wu, Tan, Xiao and Weng.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Aiewsakun P., Adriaenssens E. M., Lavigne R., Kropinski A. M., Simmonds P. (2018). Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: steps towards a unified taxonomy. J. Gen. Virol. 99, 1331–1343. doi: 10.1099/jgv.0.001110, PMID: - DOI - PMC - PubMed
-
- Alcock B. P., Huynh W., Chalil R., Smith K. W., Raphenya A. R., Wlodarski M. A., et al. (2023). CARD 2023: expanded curation, support for machine learning, resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51, D690–d699. doi: 10.1093/nar/gkac920, PMID: - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous