

Clonal competition assays identify fitness signatures in cancer progression and resistance in multiple myeloma
- PMID: 38993727
- PMCID: PMC11237348
- DOI: 10.1002/hem3.110
Clonal competition assays identify fitness signatures in cancer progression and resistance in multiple myeloma
Abstract
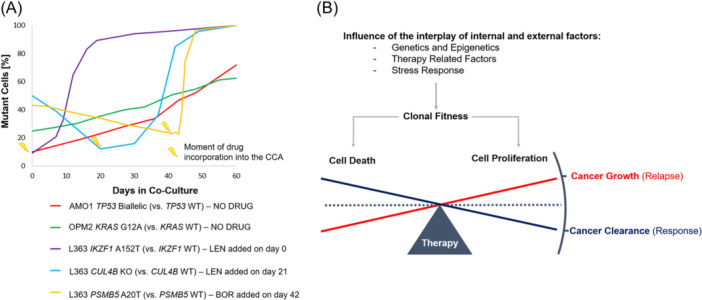
Multiple myeloma (MM) is a genetically heterogeneous disease and the management of relapses is one of the biggest clinical challenges. TP53 alterations are established high-risk markers and are included in the current disease staging criteria. KRAS is the most frequently mutated gene affecting around 20% of MM patients. Applying Clonal Competition Assays (CCA) by co-culturing color-labeled genetically modified cell models, we recently showed that mono- and biallelic alterations in TP53 transmit a fitness advantage to the cells. Here, we report a similar dynamic for two mutations in KRAS (G12A and A146T), providing a biological rationale for the high frequency of KRAS and TP53 alterations at MM relapse. Resistance mutations, on the other hand, did not endow MM cells with a general fitness advantage but rather presented a disadvantage compared to the wild-type. CUL4B KO and IKZF1 A152T transmit resistance against immunomodulatory agents, PSMB5 A20T to proteasome inhibition. However, MM cells harboring such lesions only outcompete the culture in the presence of the respective drug. To better prevent the selection of clones with the potential of inducing relapse, these results argue in favor of treatment-free breaks or a switch of the drug class given as maintenance therapy. In summary, the fitness benefit of TP53 and KRAS mutations was not treatment-related, unlike patient-derived drug resistance alterations that may only induce an advantage under treatment. CCAs are suitable models for the study of clonal evolution and competitive (dis)advantages conveyed by a specific genetic lesion of interest, and their dependence on external factors such as the treatment.
© 2024 The Author(s). HemaSphere published by John Wiley & Sons Ltd on behalf of European Hematology Association.
Conflict of interest statement
Joaquin Martinez‐Lopez and Santiago Barrio are equity shareholders of Altum Sequencing Co. The remaining authors declare no competing financial interests.
Figures
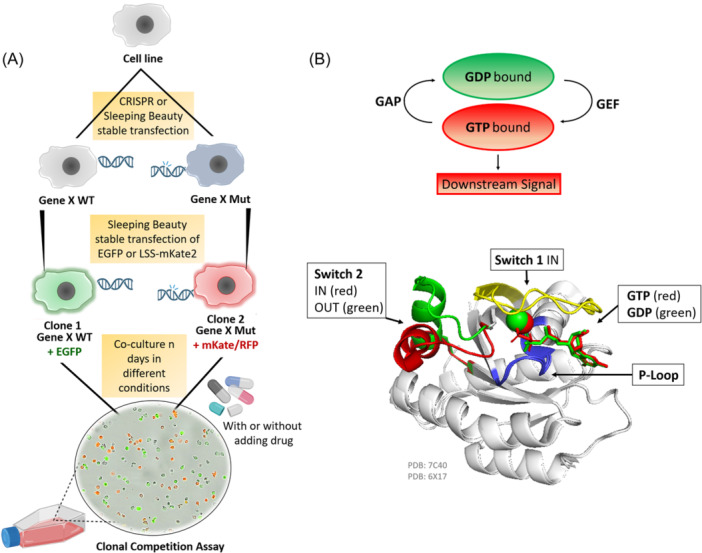
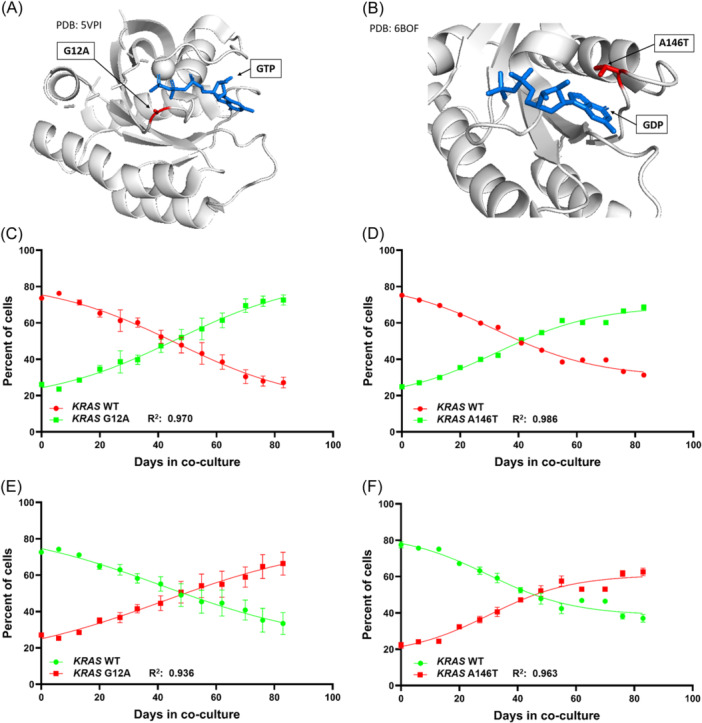
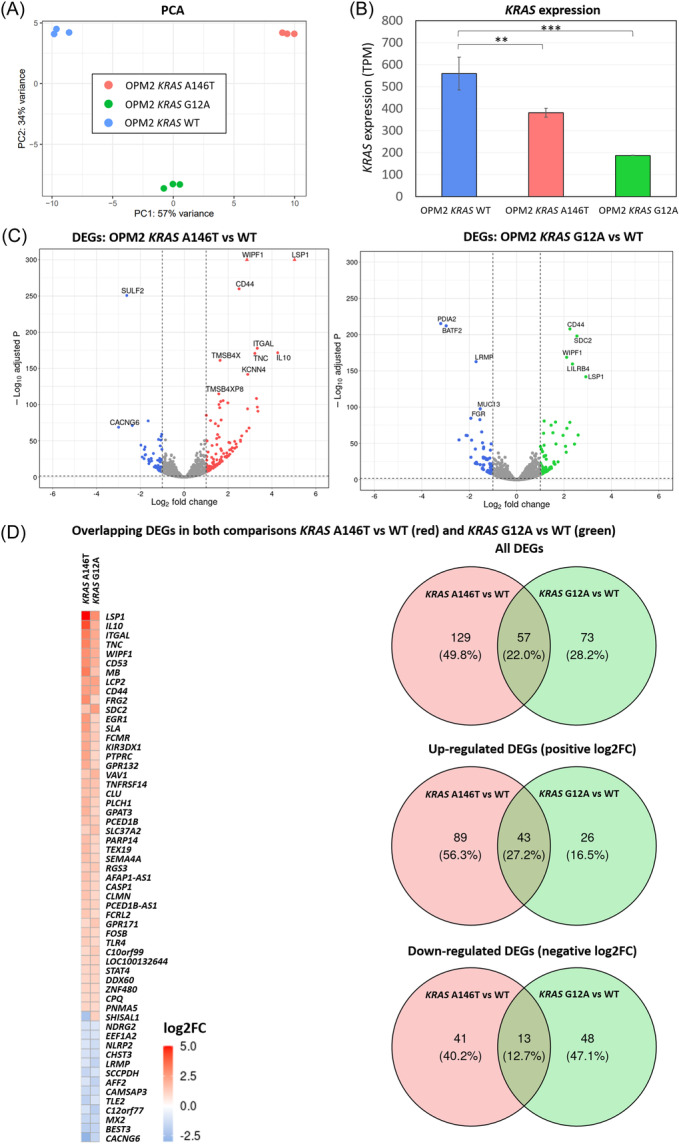
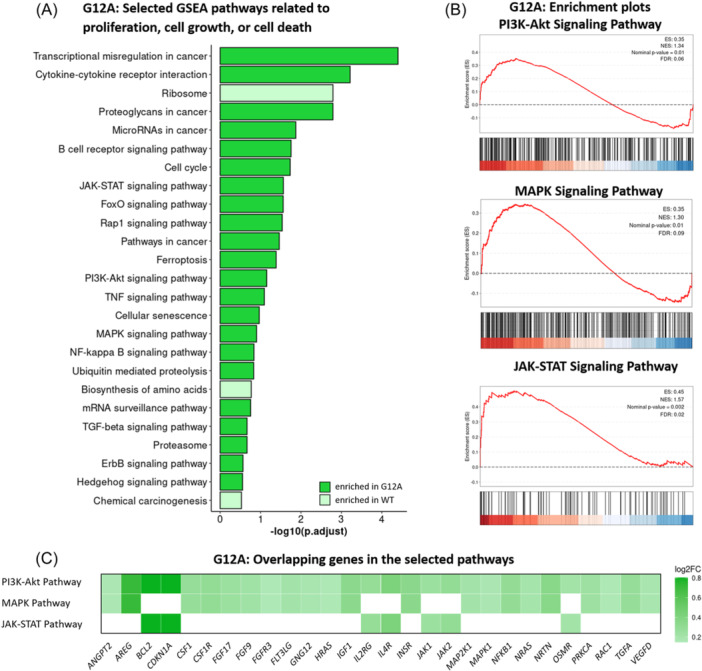
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous