

Ligand-Directed Labeling of the Adenosine A1 Receptor in Living Cells
- PMID: 38994645
- PMCID: PMC11284787
- DOI: 10.1021/acs.jmedchem.4c00835
Ligand-Directed Labeling of the Adenosine A1 Receptor in Living Cells
Abstract
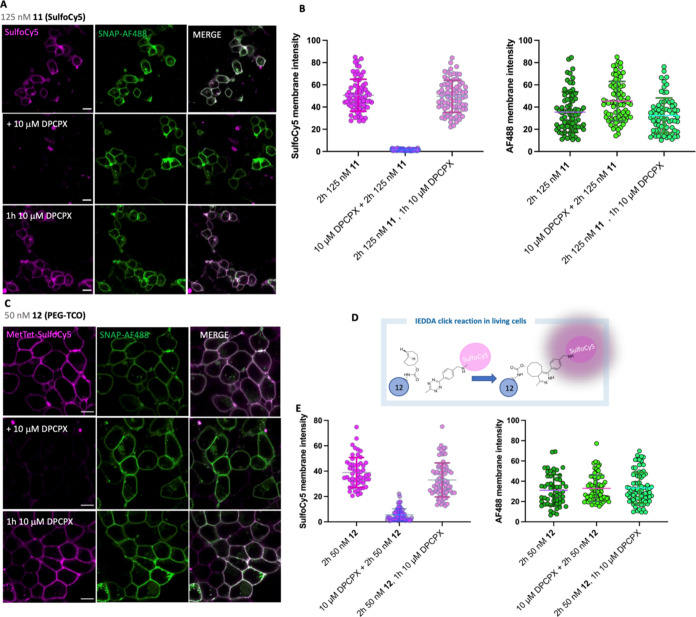
The study of protein function and dynamics in their native cellular environment is essential for progressing fundamental science. To overcome the requirement of genetic modification of the protein or the limitations of dissociable fluorescent ligands, ligand-directed (LD) chemistry has most recently emerged as a complementary, bioorthogonal approach for labeling native proteins. Here, we describe the rational design, development, and application of the first ligand-directed chemistry approach for labeling the A1AR in living cells. We pharmacologically demonstrate covalent labeling of A1AR expressed in living cells while the orthosteric binding site remains available. The probes were imaged using confocal microscopy and fluorescence correlation spectroscopy to study A1AR localization and dynamics in living cells. Additionally, the probes allowed visualization of the specific localization of A1ARs endogenously expressed in dorsal root ganglion (DRG) neurons. LD probes developed here hold promise for illuminating ligand-binding, receptor signaling, and trafficking of the A1AR in more physiologically relevant environments.
Conflict of interest statement
The authors declare no competing financial interest.
Figures









References
-
- Los G. V.; Al D.; Learish R.; McDougall M. G.; Encell L. P.; Friedman-Ohana R.; Wood M.; Vidugiris G.; Zimmerman K.; Otto P.; Klaubert D. H.; Wood K.. One Fusion Protein: Multiple Functions, Promega Notes 89, 2005; pp 2–6.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
