

Melanoma progression and prognostic models drawn from single-cell, spatial maps of benign and malignant tumors
- PMID: 38996022
- PMCID: PMC11244543
- DOI: 10.1126/sciadv.adm8206
Melanoma progression and prognostic models drawn from single-cell, spatial maps of benign and malignant tumors
Abstract
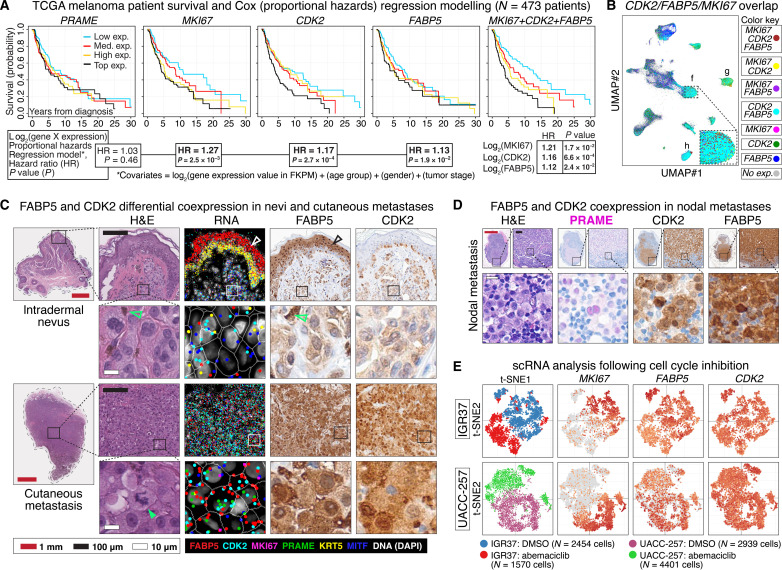
Melanoma clinical outcomes emerge from incompletely understood genetic mechanisms operating within the tumor and its microenvironment. Here, we used single-cell RNA-based spatial molecular imaging (RNA-SMI) in patient-derived archival tumors to reveal clinically relevant markers of malignancy progression and prognosis. We examined spatial gene expression of 203,472 cells inside benign and malignant melanocytic neoplasms, including melanocytic nevi and primary invasive and metastatic melanomas. Algorithmic cell clustering paired with intratumoral comparative two-dimensional analyses visualized synergistic, spatial gene signatures linking cellular proliferation, metabolism, and malignancy, validated by protein expression. Metastatic niches included up-regulation of CDK2 and FABP5, which independently predicted poor clinical outcome in 473 patients with melanoma via Cox regression analysis. More generally, our work demonstrates a framework for applying single-cell RNA-SMI technology toward identifying gene regulatory landscapes pertinent to cancer progression and patient survival.
Figures
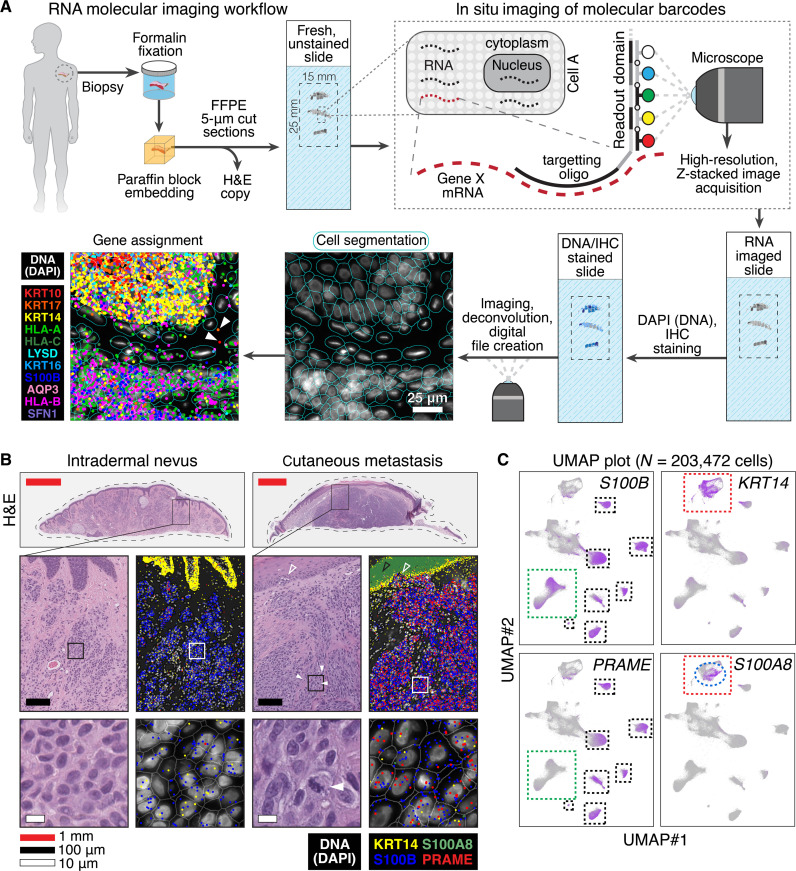
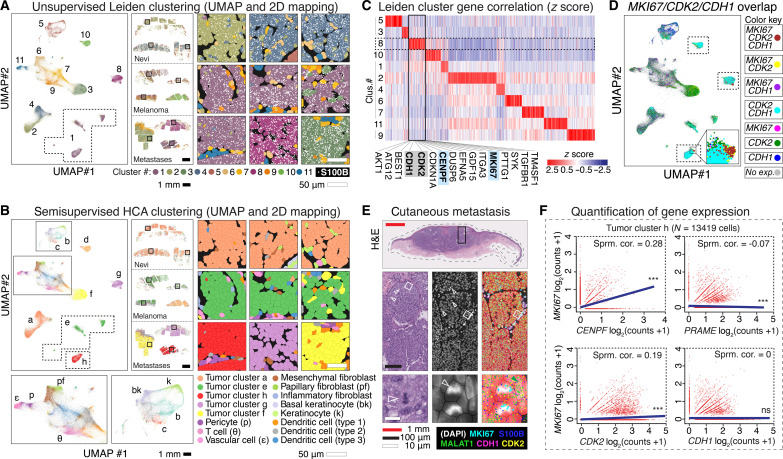
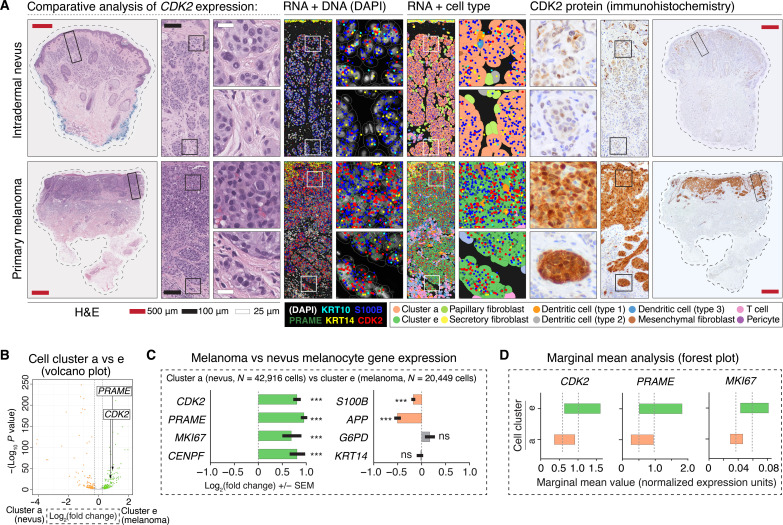
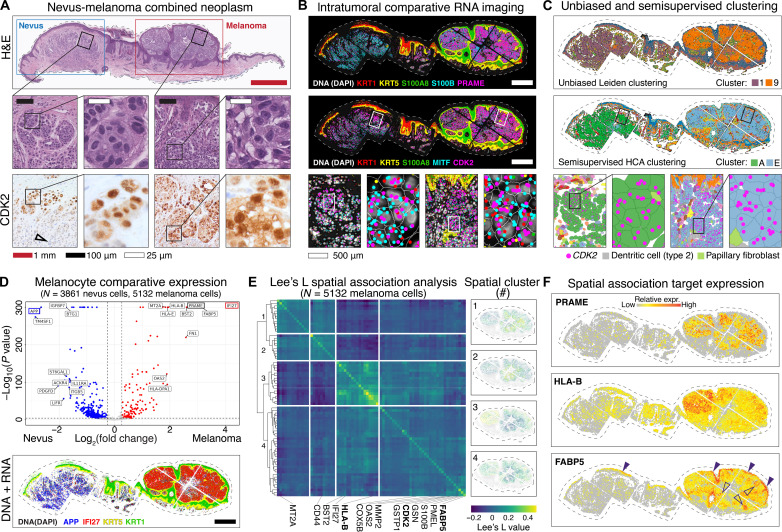
References
-
- Yeh I., Update on classification of melanocytic tumors and the role of immunohistochemistry and molecular techniques. Semin. Diagn. Pathol. 39, 248–256 (2022). - PubMed
-
- Widmer D. S., Cheng P. F., Eichhoff O. M., Belloni B. C., Zipser M. C., Schlegel N. C., Javelaud D., Mauviel A., Dummer R., Hoek K. S., Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res. 25, 343–353 (2012). - PubMed
-
- Hoek K. S., Eichhoff O. M., Schlegel N. C., Döbbeling U., Kobert N., Schaerer L., Hemmi S., Dummer R., In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 68, 650–656 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
